Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
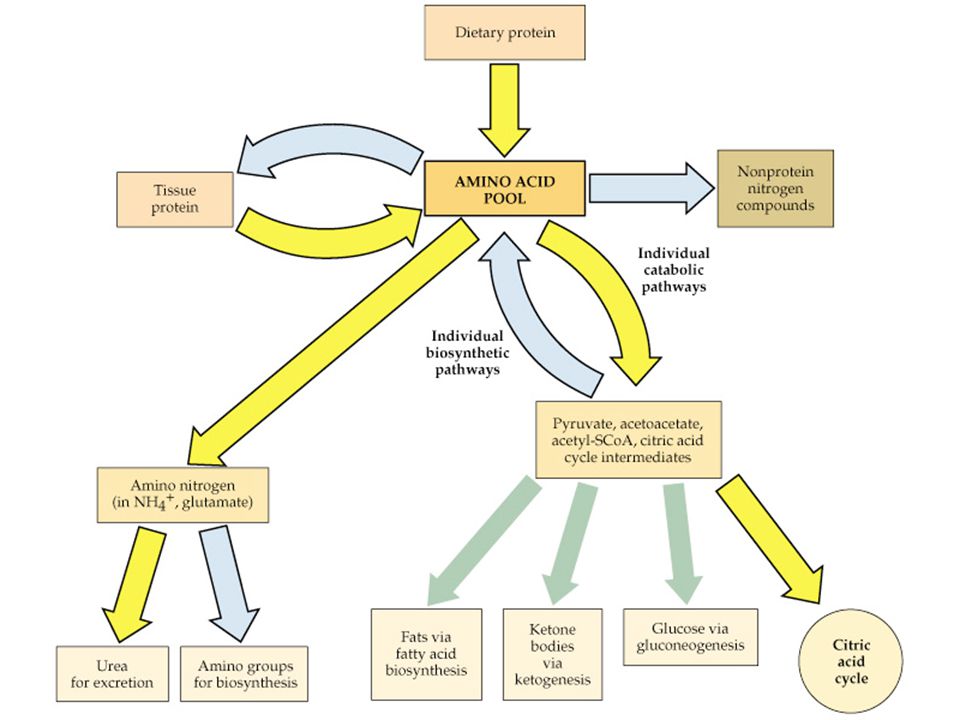
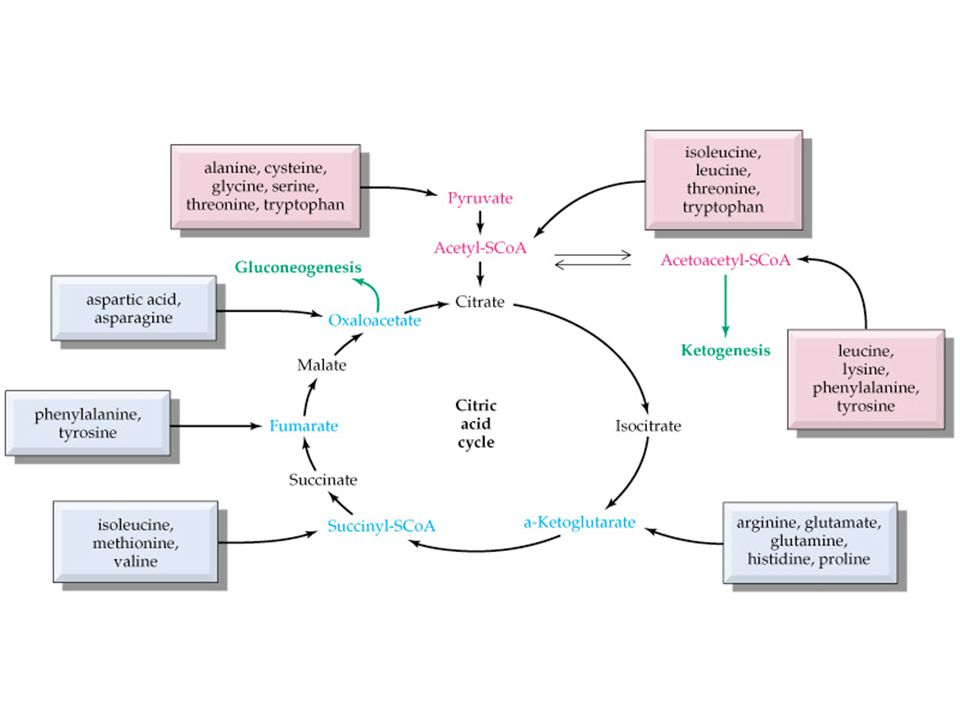
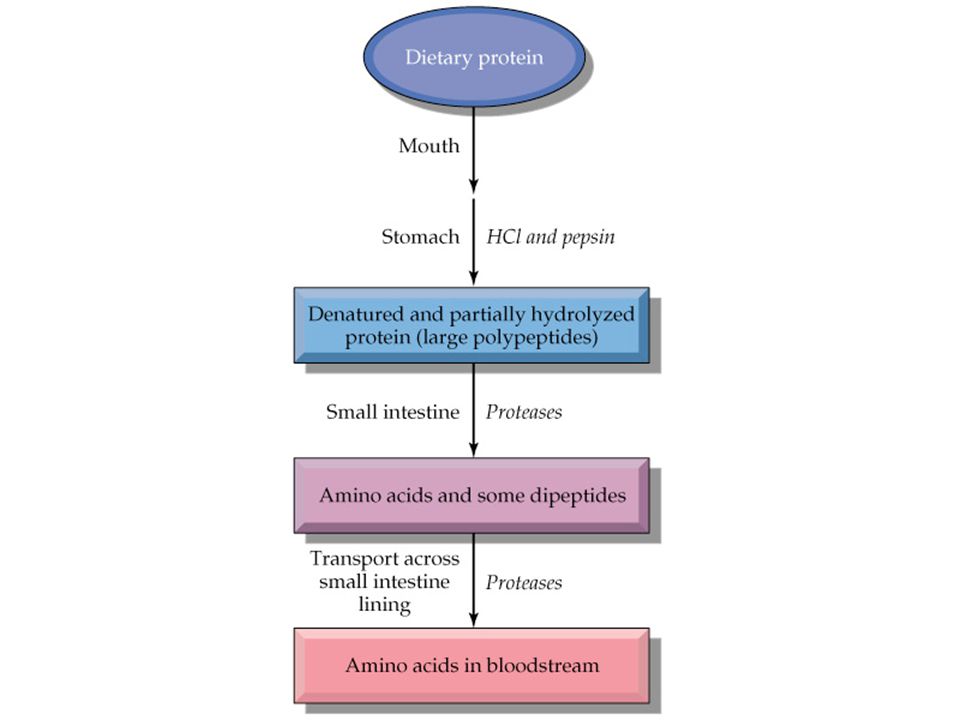
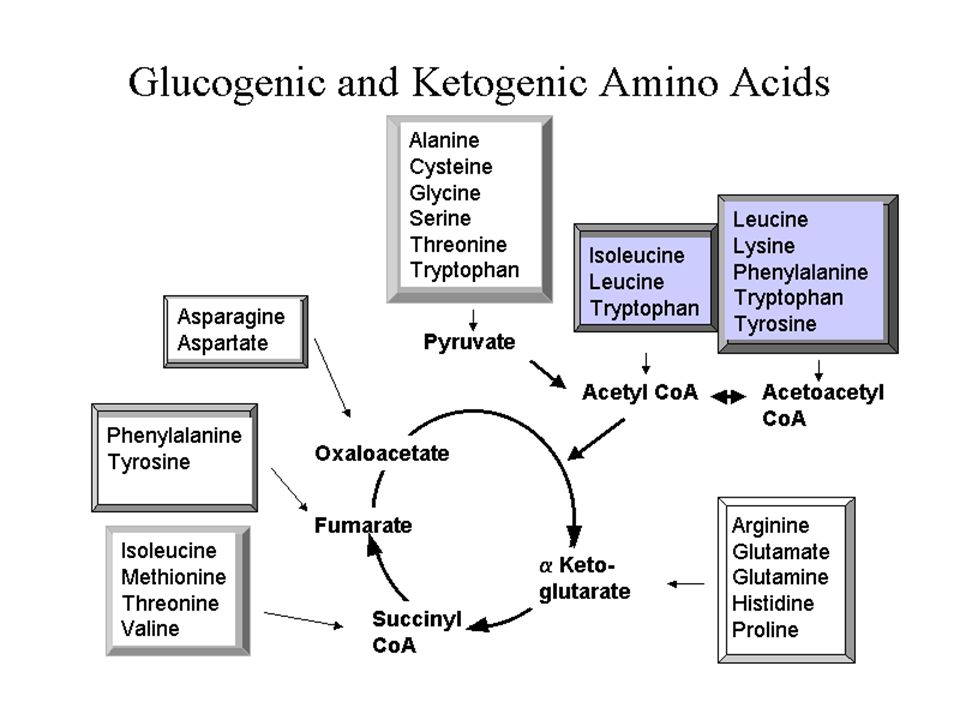
Amino asitlerin geniş spektrumu
Sitrik asit siklusu destekleyicileri Protein yapı üniteleri Peptid hormonlar(insülin,.....) Nörotransmitterler(Katekolaminler, serotonin) Nöromodulatörler(arginin, taurine) Safra asitlerinin prekürsoru(taurine) İmmun fonksiyon destekleyicileri Muskuler aktivitenin regulasyonu Nöral sinyallerin transmisyon ve kontrolu
 Nörotransmitterler(Katekolaminler, serotonin) Nöromodulatörler(arginin, taurine) Safra asitlerinin prekürsoru(taurine) İmmun fonksiyon destekleyicileri. Muskuler aktivitenin regulasyonu. Nöral sinyallerin transmisyon ve kontrolu.")
2
Hormonal sentez ve regulasyon
Besinsel transport mekanizmaları L,gament, tendon ve kemik matriks oluşumu Enzim fonksiyonları Metionin, glisin,serin Asetilkolin Glutamik asit GABA Tirozin, fenilalanin Dopamin, norepinefrin Triptofan Serotonin

8
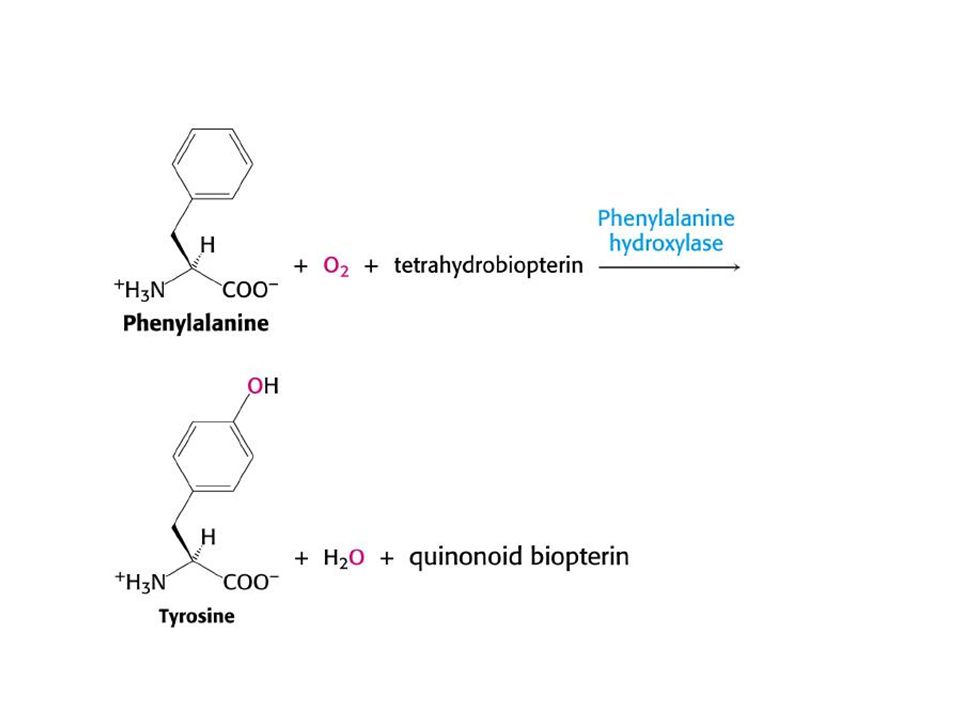
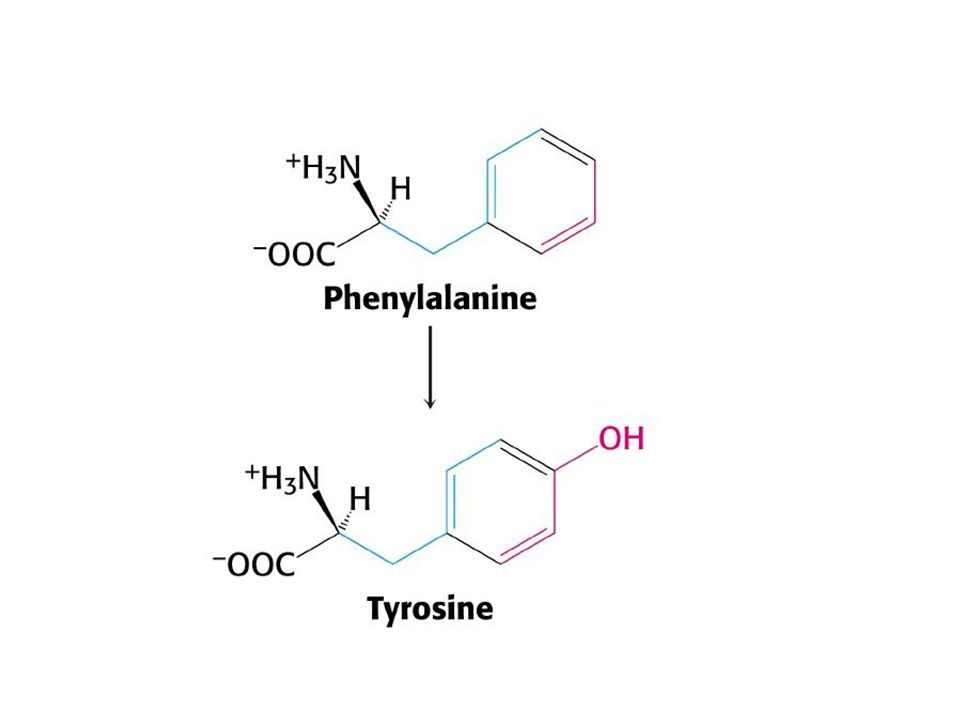
Fenilketonüri(PKU): Amino asit metabolizmasının en sık karşılaşılan hastalığı, yaklaşık15000 yenidoğan birinde görülmektedir Otozomal resesif taşınmakta İsmini idrarda yüksek miktarda bulunan fenilketondan almaktadır Buyük çapta fenilalanin hidroksilaz eksikliğine bağlı olmakla birlikte ,

9
daha nadir olarak tetrahidrobiopterin kofaktörünün veya kofaktörün indirgenmiş aktif durumda kalmasını sağlayan dihidrobiopterin redüktaz eksikliğine bağllı olarak ortaya çıkmaktadır. fenilalanin hidroksilaz kusuru tam(klasik PKU ) veya kısmi (tip ll ve lll)olabilmektedir.
 veya kısmi (tip ll ve lll)olabilmektedir.")
10
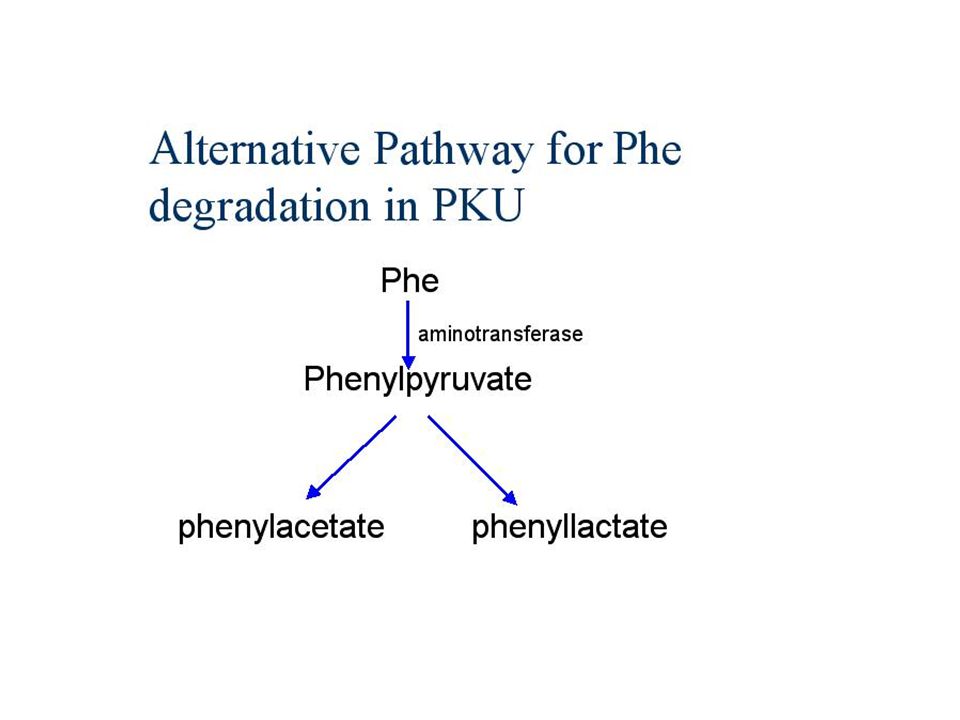
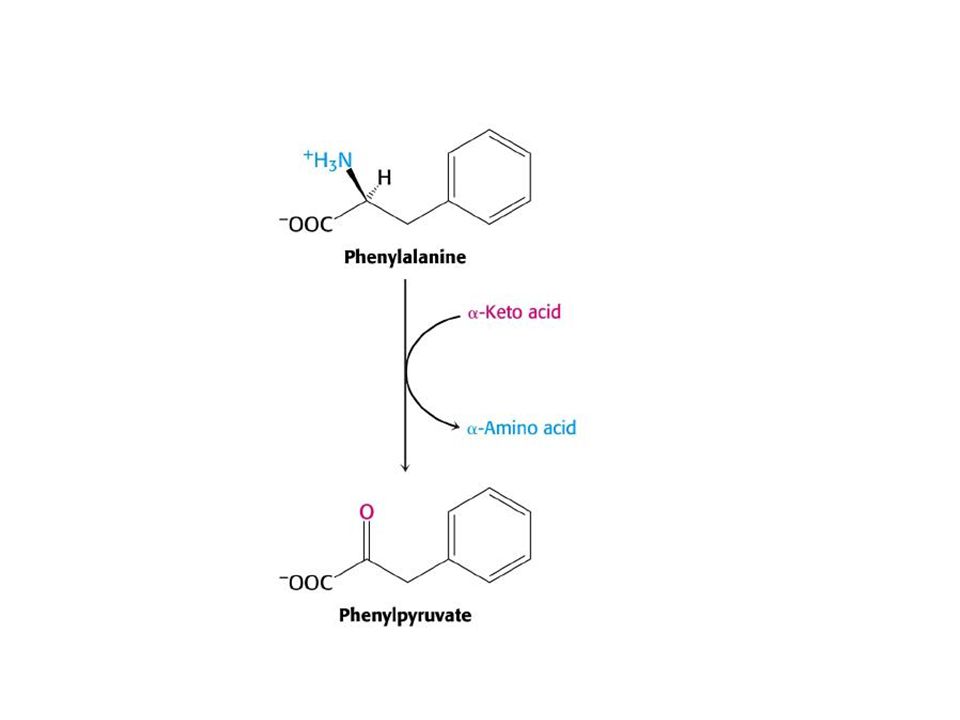
Tüm olguların yaklaşık yarısını oluşturan klasik PKU de fenilalanin hidroksilazhiç bulunmamakta ve normal şekilde katabolize olamayan fenilalanin tüm vücut sıvılatında birikmektedir. Normalde <%2 mg olan erişkin pha düzeyi , klasik tipte >%20 mg olabilmektedir Fenialanin, fenilpiruvat üzerinden katabolize olduğu için idrar ile büyük miktarda fenilpiruvat ile bu ketonun dönüştüğü fenilasetat ,fenilasetilglutamin artmaktadır.

11
Doğumda normal görünen bebeklerde kısa zaman içinde gelişme geriliği, beslenme güçlüğ ve kusma belirtileri ortaya çıkar. Artmış fenilpiruvat nedeni ile bazı bazı bebelerin idrar ve terlerinde küf kokusu olmaktadır. Fenilalanin ve metabolitlerinin birikimi beyin gelişimini bozarak zeka geriliğine yol açmaktadır. Beyinde gelişen kusurlu miyelinizasyon yüzünden sık epilepik nöbetler görülmektedir.

12
Fenilketonların beyinde serotonin enzimatik sentezini engellemelerine bağlı olarak düşük serotonin düzeyinin bu hastalardaki zeka kusurlarının nedeni olabilir Ayrıca fenilglutaminin oluşumunda fazla glutamin kullanılmasına bağlı olarak , kronik glutamin azlığının beyin hasarında doğrudan etkili olabileceği ileri sürülmektedir Deri, gözler, saçlar ve dokuların açık olması ,tirozin eksikliğinin karatteristik olduğunu düşündürmektedir.

13
Çünkü besinsel tirozin, endojen tirozin sentez eksikliğini ortadan kaldırmada yeterli görülmemektedir. Fenilketonüride erken tanı önemlidir. Dolayısıyla neonetal dönemde kan fenilalanin düzeyinin ölçülmesi önemlidir En yaygın kullanılan ve yarı kantitatif mikrobiyolojik yöntem olan Gutrie testi dir

14
Tionil alaninin basillus subtilisin üremesi üzerine olan inhibitör etkisini fenilalaninin önlemesine dayanmaktadır Fenilalaninin alkali ninhidrin ile floresan kompleks olşturmasına dayanan yöntemde neonatal tanıda sıklıkla kullanılmaktadır. PKU hastalarda, diet etkisini izlemek için yenidoğanlarda fazla miktarda atılan fenilpiruvatın, FeCl3 ile mavi-yeşil renk vermesine dayanan kalitatif idrar testinden de yaralanılır.

15
PKU tedavisi düşük proteinli besinler ile yapılmaktadır
PKU tedavisi düşük proteinli besinler ile yapılmaktadır. Geri dönüşümü olmayan beyin hasarını önlemek için düşük proteinli besin tedavisinin doğum hemen sonra başlaması ve bu tedavinin 6. Yaşa kadar devam etmesi gerekmektedir. Biopterin ve biopterin reduktaz eksikliğinin tedavisi, fenilalanin ve tirozin kısıtlılığı ile çözülmez.(sentezlenemeyen öncül bileşikler veilmeli)
")
16
Geçici neonatal hiperalaninemi:
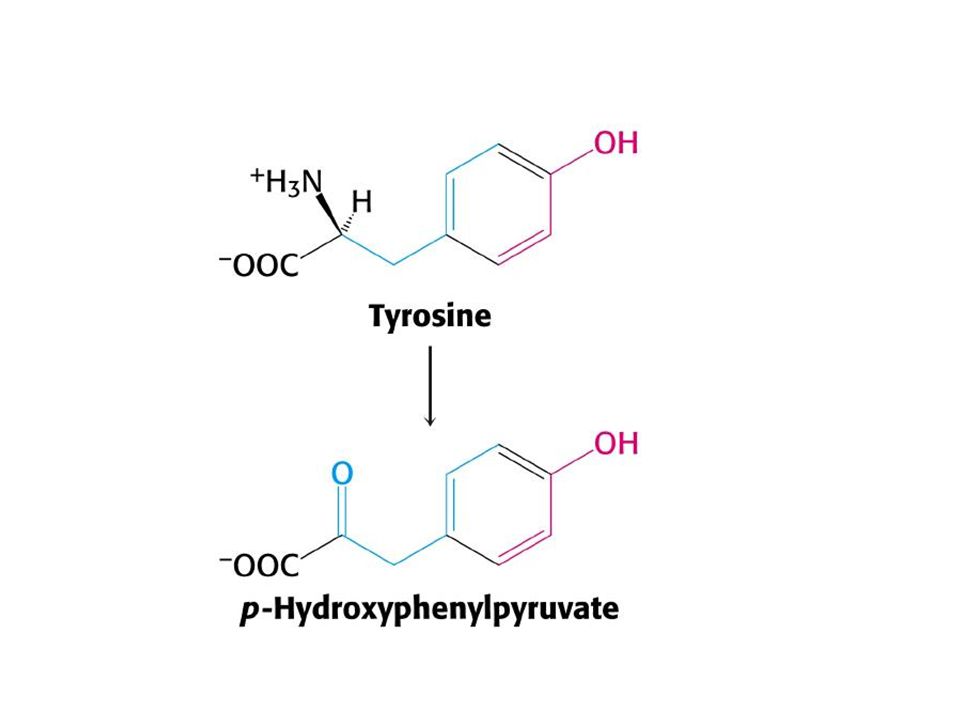
Kalıtsal bir eksiklik olmayan bu bozukluk, karaciğerin olgunlaşmasının gecikmesine bağlı olarak fenilalanin enzim sisteminin yetersizliğidir. Bebek büyükce fenilalanin düzeyi normal sınırlar içinde kalır. Tirozinemiler: Tip ll tirozinemi: tirozin transaminaz eksikliği, (okülokütanöz tirozinemi)
")
17
hipertirozinemi ve tiroziüri ile ortaya çıkar.
İdrarda n-asetiltirozin, hidroksifenil piruvat, hidroksifenil laktat ve hidroksi fenilasetat gibi tirozin metabolitleri ile tiramin metabolitlerin atılımı artmaktadır Göz ve deri lezyonları, tirozinin, intarseluler kristaleşmesine bağlı olan inflamasyon ve birçok olguda görülen zeka geriliği ile karakterize bir hastalıktır.

18
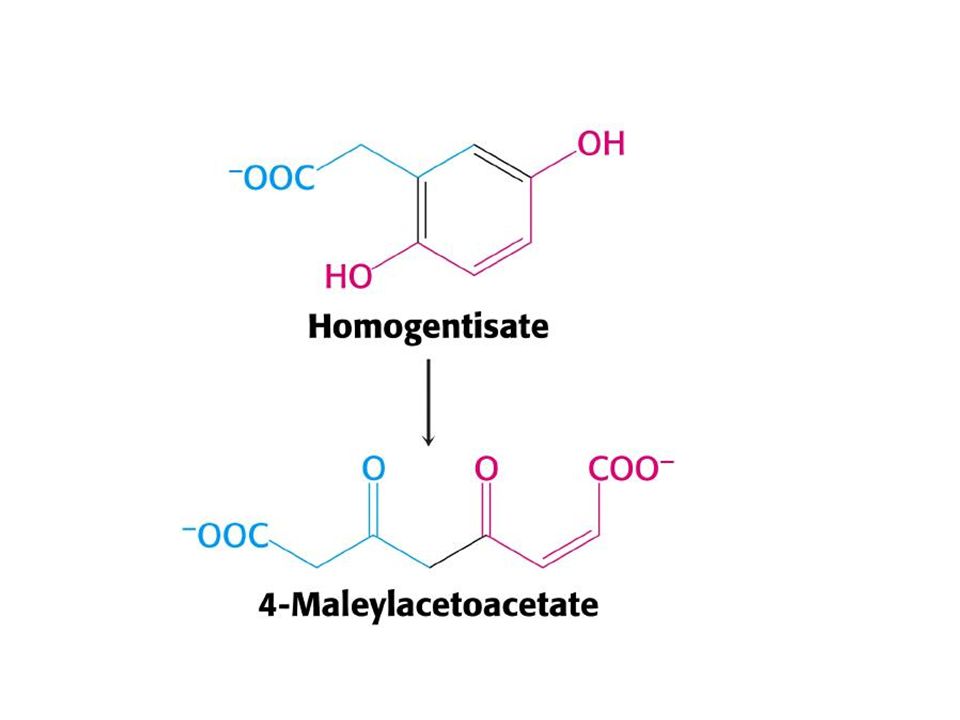
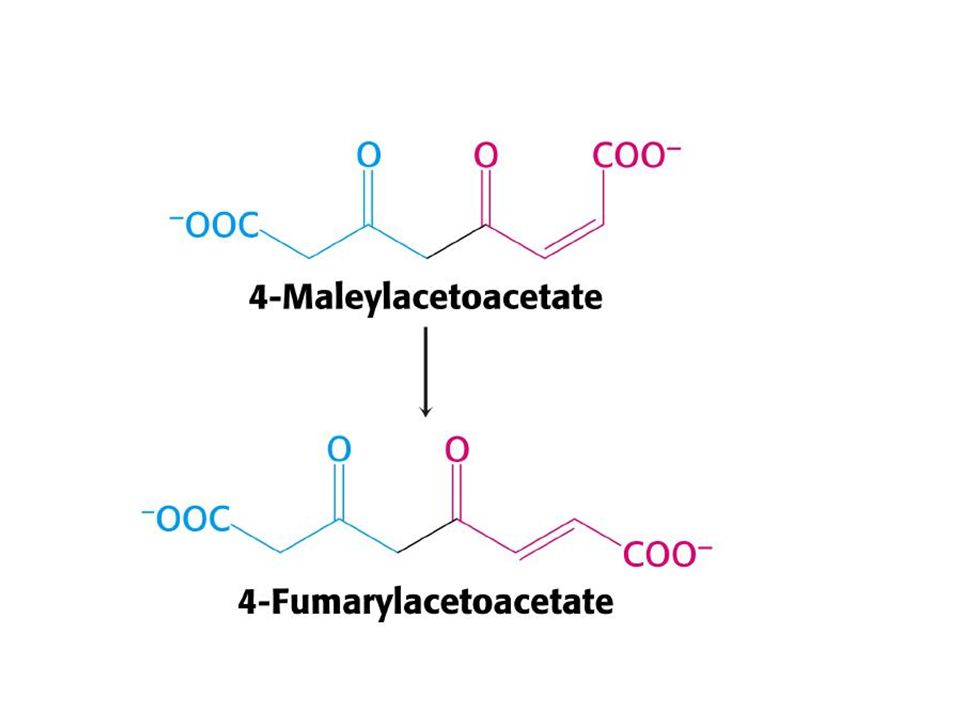
Tip l tirozinemi(hepatorenal tirozinemi).
Fumarilasetoasetat hidroksilaz eksikliğine bağlı olduğu sanılmaktadır. Karaciğer yetmezliği,renal tübüler fonksiyon bozukluğu, anemi ve D-vitaminine dirençli raşitizim ile birlikte ,idrar ile tirozin , metiyonin gibi amino asitler ve metabolitleri atılmaktadır.

19
Besinsel kısıtlama , ilerleyici karaciğer hasarını iyileştirememektedir
Neonatal tirozinemi: bebeklikte tirozinemilerin en sık görülen tipidir, özellikle prematür beblerde kc in olgunlaşmamasından dolayı serum tirozin yüksektir. geçici bir durumdur, ve hidroksi-fenilpiruvat oksidaz eksikliği sonucu ortaya çıktığı düşünülmektedir. Karaciğerin olgunlaşması ile (4-8 hafta) normal dönüş olur.
 normal dönüş olur.")
20
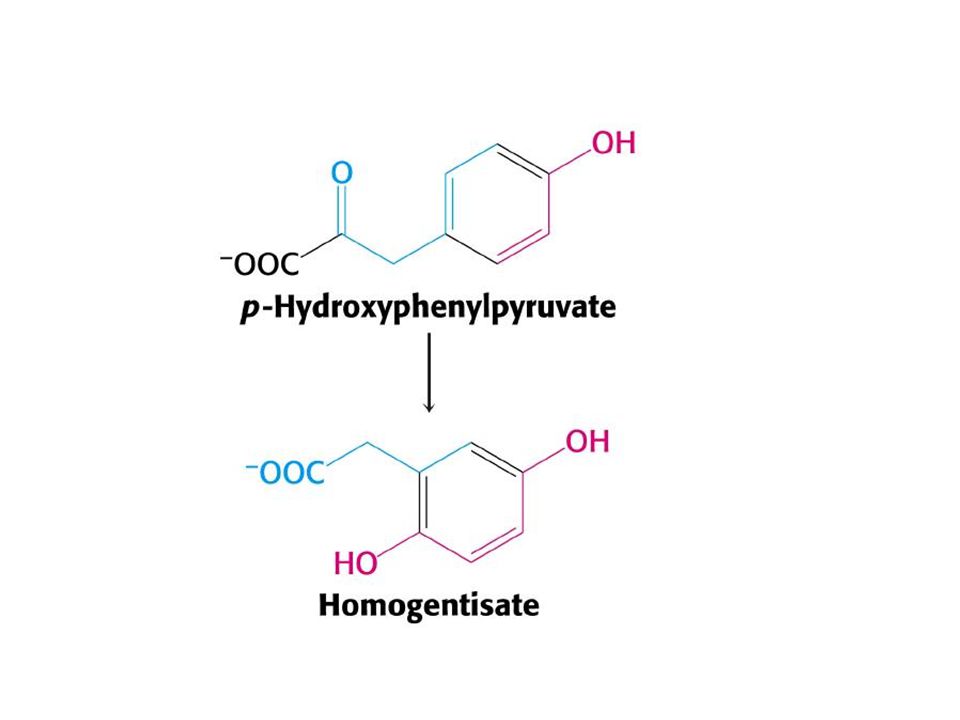
Alkaptonüri: (ilk tanımlanan kalıtsal metabolik hastalık), homogentizik asit oksidaz eksikliğine bağlı olarak homogentizik asit birikmekte ve idrarla büyük miktarda atılmaktadır. Renksiz bir hidrokinon olan bu bileşik, beklediği zaman oksidasyon ve polimerizasyon ile koyu renkli melanine benzeyen bir pigment oluşturmaktadır. Alkaptonürinin en çarpıcı klinik belirtisi idrar renginin açık havada bekletilmekle esmerleşmesidir.

21
Hastalığın geç evrelerinde oksidlenmiş homogentizik asit kıkırdak, kemikler ve diğer organlarda birikmesi sonucu görülen yaygın pigmantasyona okronozis adı verilmektedir. Özellikle erkeklerde pigment depolanması artirit gelişimine neden olmaktadır. Tirozin ve fenilalanin kısılanması yaralı olmaktadır. Ayrıca homogentisik asit oksidazın maksimum aktivitesi için c vitamini de verilmektedir.

22
Albinizim: Melanin sentezindeki kalıtsal hatalara bağlı olarakorta bir sendrom.
Tirrozinaz enziminin(melanositlerde tirozinin melanine dönüşümünü sağlayan bakır içeren bir enzim)yokluğu veya yetersizliğine bağlı olarak deri, saç ve gözlerde pigmantasyon azalmaktadır. Deride pigment azlığı , albinoların güneşe duyarlılığını artırmakta ve sıklıkla deri kanserlerine yol açmaktadır.İris pigmantasyonunun azlması, fotofobiye neden olmaktadır.
yokluğu veya yetersizliğine bağlı olarak deri, saç ve gözlerde pigmantasyon azalmaktadır. Deride pigment azlığı , albinoların güneşe duyarlılığını artırmakta ve sıklıkla deri kanserlerine yol açmaktadır.İris pigmantasyonunun azlması, fotofobiye neden olmaktadır.")
Benzer bir sunumlar