Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
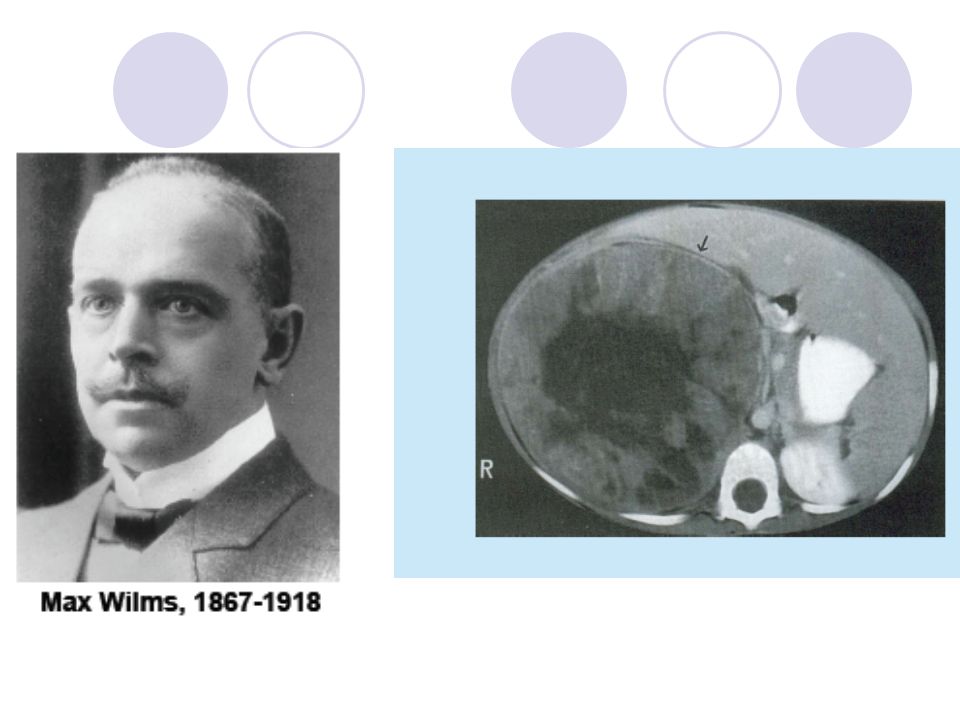
Solid Tümörler Uzm. Dr. H. Murat MUTUŞ

3
Wilms Tümörü Nefroblastoma da denilir
Çocuklarda en sık görülen solid tümörlerden biridir 15 yaş altında 8/106 oranında görülür. Görülme sıklığı coğrafik bölgelere göre değişir Yaş, cinsiyet ve bilateralite: Erkek/ kız oranı=1/1,1 Olguların başvuru yaşı 3,5-4 yaş civarıdır Bilateral Wilms tümörü %7 oranında görülür

4
Wilms Tümörü Birlikte Görülen Anomaliler
Aniridi, Hemihipertrofi İnmemiş testis, Hipospadias Male pseudohermafroditizm Renal hipoplazi Renal ektopi Duplikasyon anomalileri Atnalı böbrek

5
Wilms Tümörü Birlikte görülen sendromlar
WAGR sendromu Wilms Tümörü, aniridi, genitoüriner malformasyon, mental retardasyon Wiedemann- Beckwith sendromu (WBS) Makroglossi, organomegali, hemihipertrofi, karın duvarı defektleri, hipoglisemi, kulak kepçesi deformitesi Drash sendromu Pseudohermafrotidizm, dejeneratif böbrek hastalıkları Klippel-Trenaunay sendromu Perlmann sendromu Spesifik sitogenetik anormallikler
 Makroglossi, organomegali, hemihipertrofi, karın duvarı defektleri, hipoglisemi, kulak kepçesi deformitesi. Drash sendromu. Pseudohermafrotidizm, dejeneratif böbrek hastalıkları. Klippel-Trenaunay sendromu. Perlmann sendromu. Spesifik sitogenetik anormallikler.")
6
Wiedemann- Beckwith sendromu (WBS)
Makroglossi Organomegali Hemihipertrofi Karın duvarı defektleri Hipoglisemi Kulak kepçesi deformitesi

7
Wilms Tümörü Aniridia Hemihipertrofi:
1000 kez daha fazla Wilms olma şansına sahiptir Aniridili tüm çocuklar ilk 2 yaşta her 3 ayda bir, 4 yaşına kadar 6 ayda bir fizik muayane ve abdominal ultrasound kontrolünden geçmelidir. Hemihipertrofi: Normale göre 1000 kat daha fazla risk altındadırlar. Bu olgular da yukarıdakine benze şekilde dikkatli izlenmelidir

8
Wilms Tümörü Genitoüriner anomaliler Familial Wilms tümörü
İnmemiş testis ve hipospadias populasyonda sırasıyla %0,7 ve %1,0 insidansa sahip iken, Wilms tümörlü olgularda %5,2 olmaktadır. Bilateral tümör görülme olasılığı bu tür genitoüriner anomalisi olanlarda daha fazladır (%25). Familial Wilms tümörü Wilms tümöründe aile öyküsü %1,5 oranındadır Familial olgularda da Bilateralite yüksek orandadır (%16)
. Familial Wilms tümörü. Wilms tümöründe aile öyküsü %1,5 oranındadır. Familial olgularda da Bilateralite yüksek orandadır (%16)")
10
Tablo. NWTS grubun 1980-99 yıllarında saptanan Wilms Tümörlü
hastalardaki konjenital anomaliler ve sendromlar Anomali, Sendromlar Hasta sayısı Prevalans (%0) Wilms Tümörü, aniridi, genitoüriner malformasyon, mental retardasyon (WAGR)sendromu 52 7,5 Denys-Drash sendromu 28 4,0 Beckwith-Wiedeman sendr. 74 10,7 Sporadik hemihipertrofi 171 25,1 İnmemiş testis (erkeklerde) 49 46,6 Hipospadias (erkeklerde) 64 20,0
 Wilms Tümörü, aniridi, genitoüriner malformasyon, mental retardasyon (WAGR)sendromu ,5. Denys-Drash sendromu ,0. Beckwith-Wiedeman sendr ,7. Sporadik hemihipertrofi ,1. İnmemiş testis (erkeklerde) ,6. Hipospadias (erkeklerde) ,0.")
11
Moleküler Genetik: Wilms tümör geni
WT-1 geni; bir tümör supressor geni olup11. kromozomun p13 bölgesindedir (11p13). Bu gen WAGR sendromlu (Wilms tümörü, aniridi, genitoüriner anormallikler, ve mental retardasyon) çocukların tümünde delesyona uğramıştır. Oysa sporadik Wilms tümörlülerde %5-10 oranında delesyon saptanmıştır. WT-2 geni (11p15) de bir supressor gendir. Diğer genetik anormallikler saptanmıştır. Örneğin p53 supressor geni Wilms tümörünün agressif formunda rol oynar.
. Bu gen WAGR sendromlu (Wilms tümörü, aniridi, genitoüriner anormallikler, ve mental retardasyon) çocukların tümünde delesyona uğramıştır. Oysa sporadik Wilms tümörlülerde %5-10 oranında delesyon saptanmıştır. WT-2 geni (11p15) de bir supressor gendir. Diğer genetik anormallikler saptanmıştır. Örneğin p53 supressor geni Wilms tümörünün agressif formunda rol oynar.")
13
Klinik / Semptomlar Abdominal kitle Hipertansiyon Hematüri
genellikle ilk bulgudur Hipertansiyon Hematüri

14
Klinik / Bulgular Büyük, düzgün ve yumuşak bir kitle palpe edilebilir
Lezyon genellikle hassas değildir ve relatif olarak immobildir Genellikle orta hattı geçmez Bazen abdominal ağrı,ateş ve anemi intraperitoneal veya subkapsüler kanama ile tümör nekrozunun bir sonucudur. Bu yüzden nöroblastomdan ayırt etmek zordur İntravenöz uzanım kardiak murmur, hepatosplenomegali, asit, varikosel ve gonadal metastaz Genellikle akciğere metastaz yapar

15
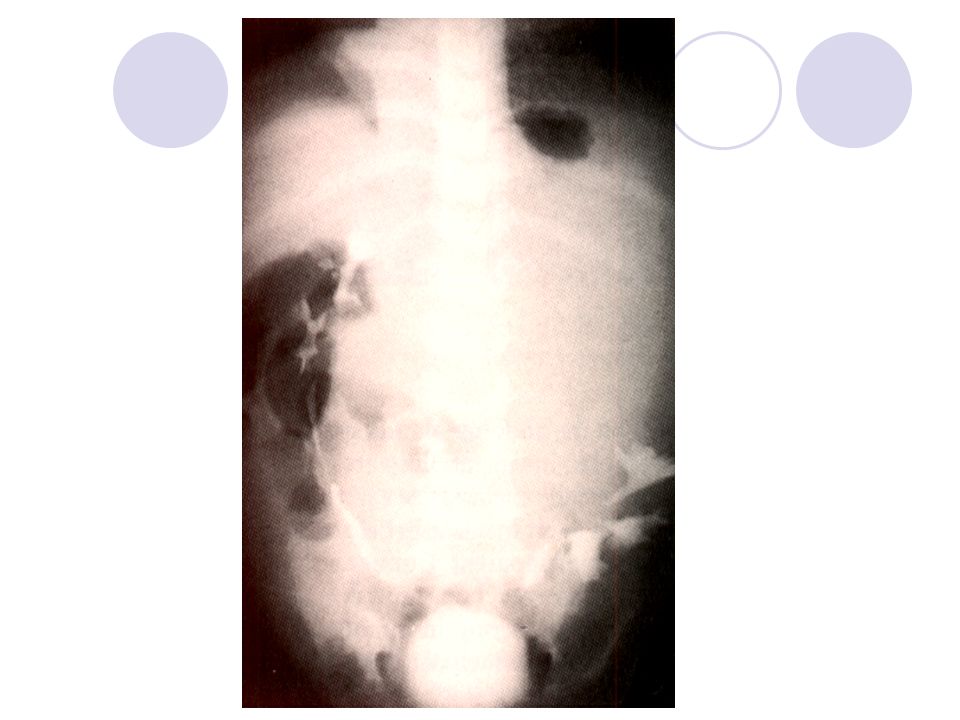
TANI / Diagnostik Görüntüleme
Sırasıyla; akciğer grafisi abdominal düz radiografileri takiben USG ve CT rutin ve MRGgerektiğinde yapılır Kitlenin natürünü ve orijin aldığı organı, Kontralateral renal dokunun durumu, Bilateral hastalığın varlığını, VCI ve renal venin açıklığını Uzak metastaz varlığını saptamak.

16
IVP

17
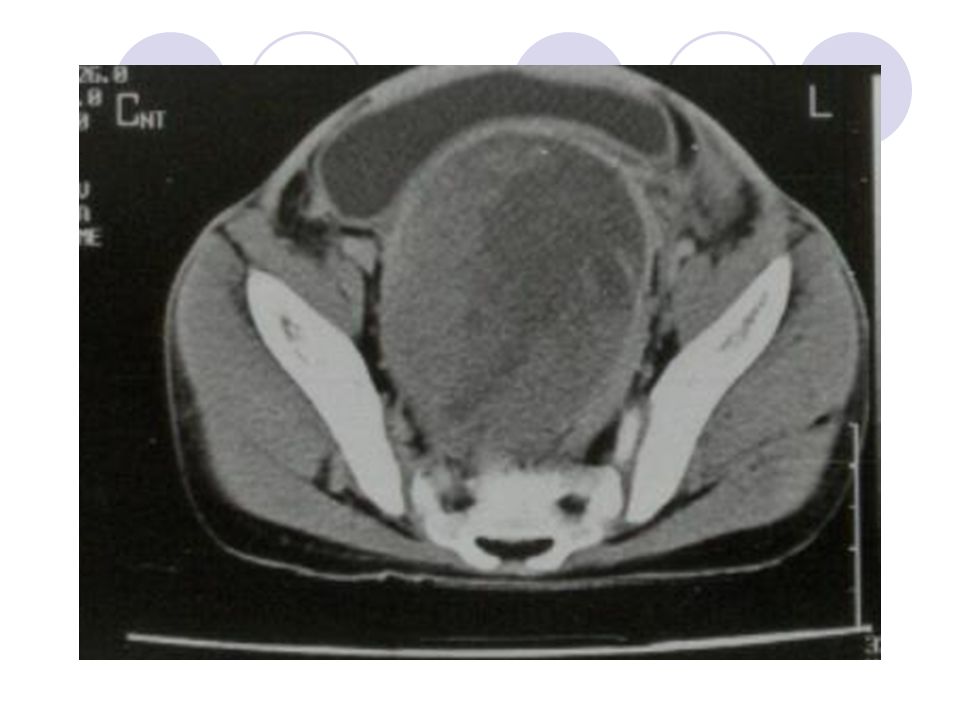
USG

18
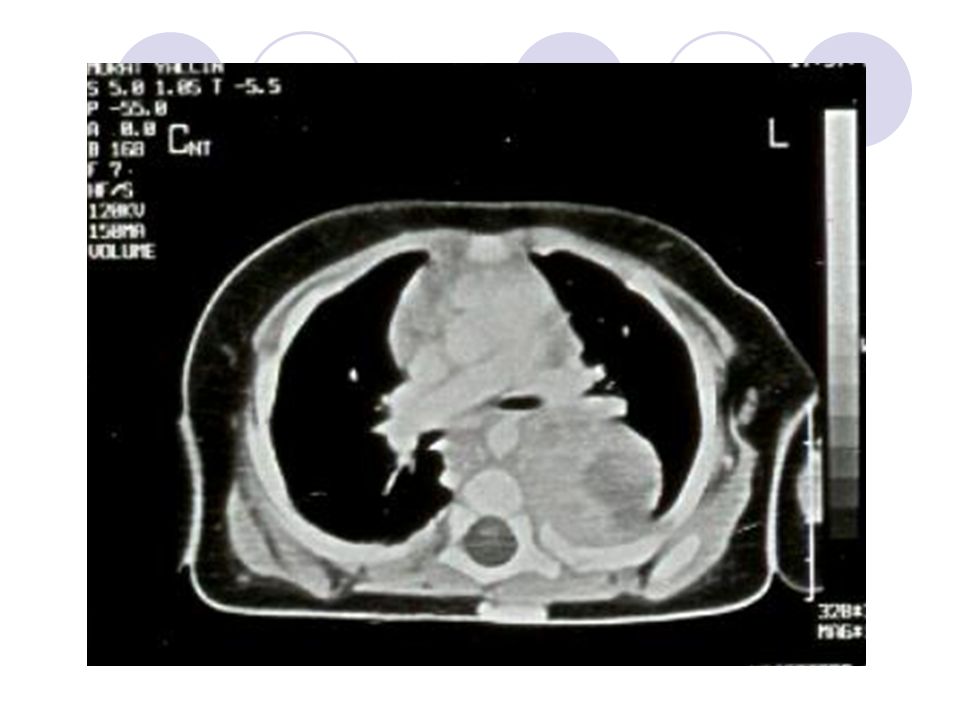
CT

19
Metastaz

20
Blastomatozis Wilms

21
Wilms + Adenopati

22
Wilms + Adenopati + Kapsül İnvazyonu

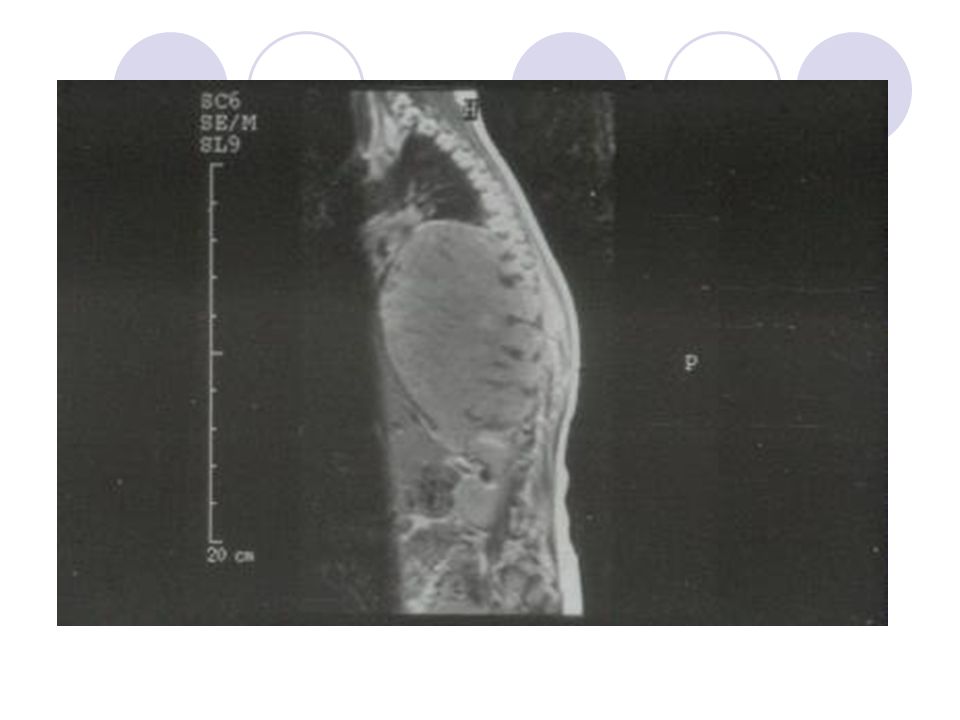
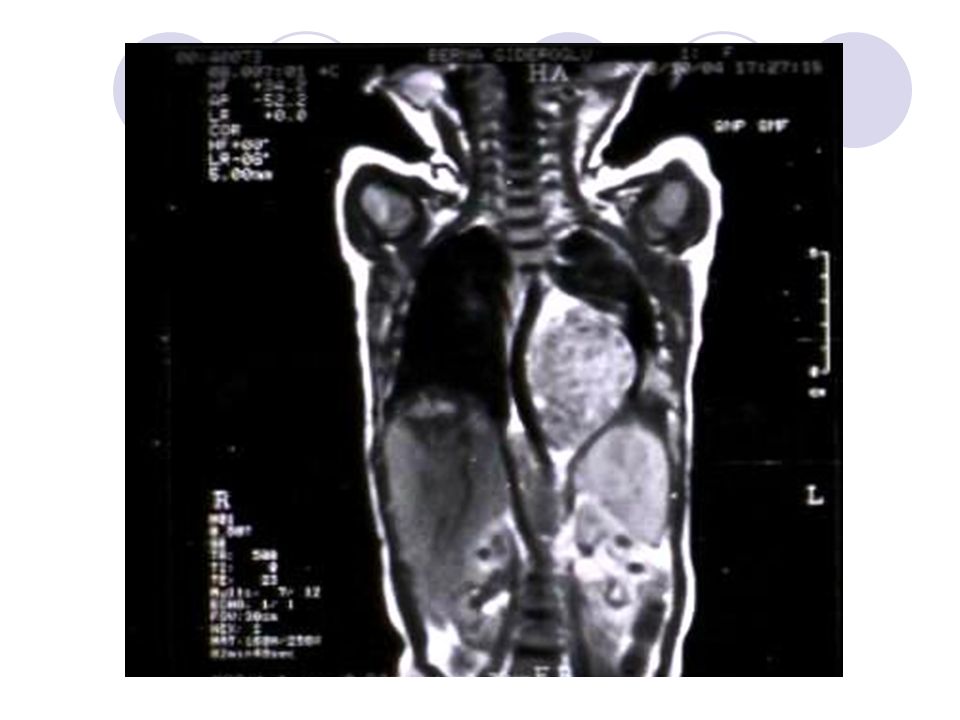
23
Bilateral Wilms Tümörü -MRI

24
Bilateral Wilms

25
Bilateral Wilms

26
Patoloji Blastemal, stromal veya epitelial elementlerden oluşur.
Herhangi hücre tipi dominant olabileceği gibi, mixed doku paterni de gözlenebilir Histopatolojik olarak; “favorable ve unfavorable histoloji” en önemli prognostik faktördür

27
Patoloji Nefroblastomatosis (Nefrojenik artık)
Premalign bir lezyon olarak kabul edilir Histolojik olarak persistan metanefrik doku söz konusudur Wilms tümörü ile açık bir ilişkisi vardır Bu durum postmortem incelemelerde %1 oranında saptanırken, Wilms tümörlü olgularda %25-40 görülür Bu da daha sonraki dönemlerde kontralateral WT riskini artıran en önemli bulgusudur

28
Nefrojenik kalıntı

29
Wilms T-NWTS-5 Evreleme Sistemi
Evre I: Tümör böbrekte sınırlı ve tamamen rezeke edilmekte - Renal kapsül intakt Evre II: Tümör böbrek ötesine uzanmakta ama tamamen çıkarılabilir Evre III: Residual nonhematogen tümör vardır ve abdomende sınırlıdır Evre IV: Abdominopelvik bölge dışına hematojen metastaz veya lenf nodu matastazı vardır. Evre V: Tanı anında bilateral böbrek tutulumu vardır.

30
Evre 1 Tümör böbrekte sınırlı ve tamamen rezeke edilmekte - Renal kapsül intakt

31
Evre 2 Tümör böbrek ötesine uzanmakta ama tamamen çıkarılabilir

32
Evre 3 Residüel non-hematogen tümör vardır ve abdomende sınırlıdır

33
Evre 4 Abdominopelvik bölge dışına hematojen metastaz veya lenf nodu matastazı vardır

34
Tanı anında bilateral böbrek tutulum
Evre 5 Tanı anında bilateral böbrek tutulum

35
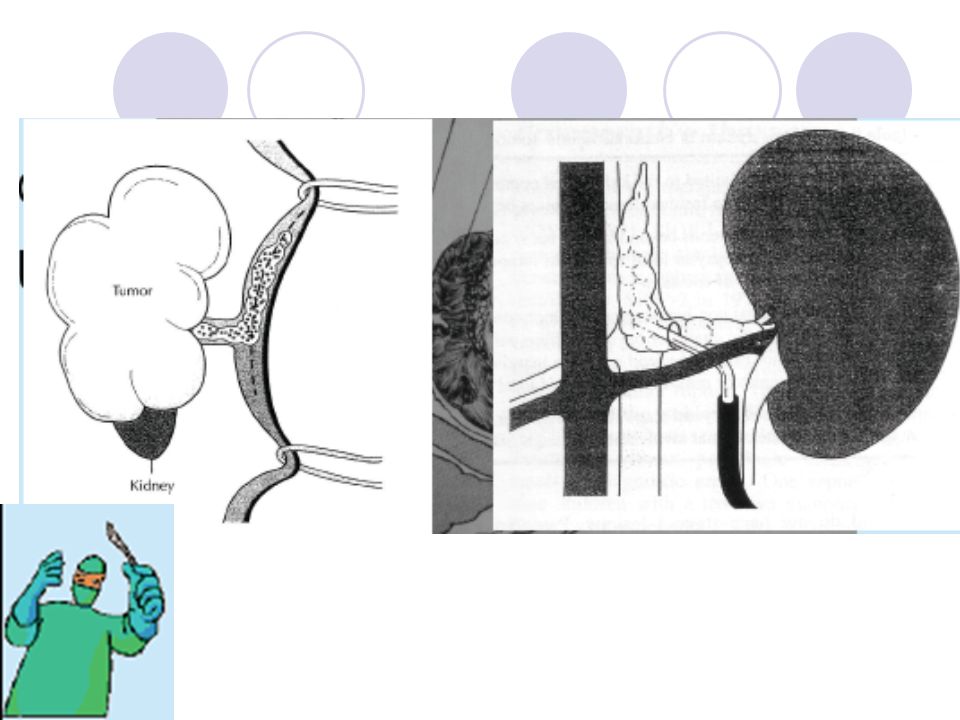
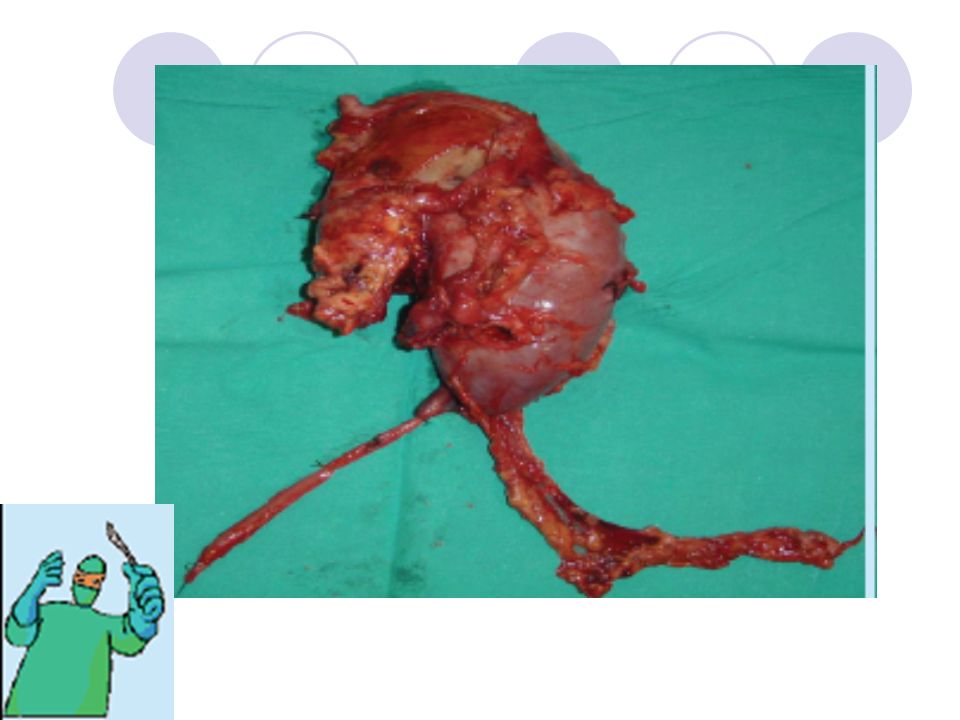
Cerrahi Tedavi Cerrahın iki temel sorumluluğu vardır:
Tüm viable tümörün komplet rezeksiyonunu sağlamak, Doğru evreleme bilgilerini sağlamaktır. Genellikle çoğu olguda unilateral radikal nefrektomi yapılır. Venöz tutulum ve bilateral Wilms tümörü varlığının araştırılması ve varsa bunlara spesifik cerrahi prosedürlerin gerçekleştirilmesi gerekir. Radyasyon ve Kemoterapi uygulamaları zaman içinde sırasıyla gerçekleştirilen “National Wilms’ Tumor Study-1,2,3,4,5” adlı klinik çalışmalar ile devamlı olarak geliştirilmektedir. Bu tedavi modaliteleri de cerrahiye ek olarak protokollere bağlı olarak kullanılmaktadır.

38
CLEAR CELL SARCOMA Çocuklarda kemiğe metastaz yapan renal tümörü olarak da bilinir. UH ye sahip WT’ne benzerler. %25 inde kemik metastazı yapar. Önemli histolojik özellikleri; tümörde fibrovasküler septasyon oluşumu ile birlikte vasküler arkus formasyonu ile tümör hücreleri kord veya kolonlara bölünmesidir. Multisentrik veya bilateral clear cell sarkom bildirilmemiştir. Tedavide tüm hastalara nefrektomi, radyoterapi ve kemoterapi standart uygulanır. Kemik metastazı varsa uzun süreli sürvey oldukça düşüktür (%16).
.")
39
RHABDOID TÜMÖRLER Nadir görülür ama çok fatal seyreder. Renal medullada nöral krest orijinli hücrelerden köken aldığı düşünülen bir tümördür. Renal tümörlerin %2 sini oluştururlar. Bu olgularda hiperkalsemi gözlenebilir. CT de WT den ayırıcı özellikler olabileceği bildirilmiştir. Tüm olgular unilateraldir. Metastaz çeşitli yerlere olabilir ama en sık pulmoner metastaz söz konusudur. Tedavide cerrahi,kemoterapi ve radyoterapi uygulanmakta olup sonuçlar pek yüz güldürücü değildir

40
RENAL ADENOCARCINOMA Oldukça nadir görülen bir tümördür. Çoğunlukla geç çocukluk ve adolesan dönemde görülür. Klasik olarak abdominal ağrı, hematüri ve flankta kitle triadı erişkinde klasik bulgular olduğu halde çocukluk ve adolesan dönemlerinde sık değildir. En sık akciğer, karaciğer ve kemiğe metastaz yaparlar. Prognoz klinikopatolojik evreye göre değişir. Önerilen tedavi, radikal nefrektomi+rejional lenfadenektomidir.Beklenen sürvey oranı ortalama %50 dir. Kemoterapi ve radyoterapi (metastatik lezyonlara) de diğer tedavilerdir.
 de diğer tedavilerdir.")
41
KONJENİTAL MEZOBLASTİK NEFROMA
Yaşamın ilk 3 ayında en sık görülen renal tümördür 4. aydan itibaren ise Wilms tümörü en sıktır Pediatrik renal tümörlerin % 5’idir %90 dan fazlası ilk 1 yaş içinde görülür Erkek çocuklarda 2 kat daha fazladır. İlk başvuru yaşı aydır

42
KONJENİTAL MEZOBLASTİK NEFROMA
Genellikle karında palpe edilen kitle ile başvururlar Hematüri az da olsa görülebilir Prenatal olarak da saptanabilir Bu olgularda polihidramnioz vardır Preterm eylem, düşük doğum ağırlığı ve hidrops fetalis de söz konusu olabilir.

43
KONJENİTAL MEZOBLASTİK NEFROMA
Bilateral lezyon bildirilmemiştir. Tedavinin en önemli parçası cerrahi eksizyondur. %95’i sadece cerrahi ile tam kür sağlanabirken, %5 olguda rekürrens görülebilir Bu açıdan cerrahi sonrası USG ile izlem önemlidir. Relatif olarak benign karekterli olan bu tümör bazen agresif özellikler de gösterebilmektedir.

44
KONJENİTAL MEZOBLASTİK NEFROMA

45
KONJENİTAL MEZOBLASTİK NEFROMA

46
KONJENİTAL MEZOBLASTİK NEFROMA
Longitudinal section of kidney showing large blood clot occupying upper and middle poles with thinned cortex

47
KONJENİTAL MEZOBLASTİK NEFROMA
Low-power photomicrograph showing cystic area, large fibrous strand, and innumerable small, round cells in renal cortex typical of mesoblastic nephroma

48
Multikistik nefroma

49
Bilateral Multikistik Nefroma

50
Lösemi Renal Tutulum

51
Nöroblastoma

52
Nöroblastoma Çocukluk çağında lenfomalar ve santral sinir sistemi tümörlerinden sonra en sık görülen solid tümörüdür Nöral krest hücrelerinden; sempatik sinir sistemi gangliyonlarını oluşturan sempatoganglionlardan ve adrenal medulladan gelişir

53
Nöroblastoma İnsidans: Irklara göre insidans değişebilir.
Asya’da 7-12 /1 milyon/yıl’dır Çocukluk çağı kanserlerinin % 6-10’unu oluşturur. %30 u ilk 1 yaşta , %50 si 1-4 yaş arasında, ve %5 i 10 yaştan sonra görülür. Erkek/Kız = 1,3/1 olup erkeklerde bir miktar daha fazla görülür.

54
Nöroblastoma Etyopatogenez: Etyolojisi bilinmiyor.
Çoğunlukla sporadiktir. Nadiren familiyal olgular tanımlanmıştır (Sıklıkla multiple primer tümörler şeklindedir). Erken fötal yaşam esnasında paravertebral ganglionlardan adrenal medulla içine nöroblast migrasyonu oluşur.Neoplasm gelişince, vasküler ve komşu yapılara invazyon ile lokal yayılımı gelişir.
. Erken fötal yaşam esnasında paravertebral ganglionlardan adrenal medulla içine nöroblast migrasyonu oluşur.Neoplasm gelişince, vasküler ve komşu yapılara invazyon ile lokal yayılımı gelişir.")
55
Nöroblastoma Metastazlar:
Lenf nodu, kemik, kemik iliği, karaciğer ve deriye olur. 4S evreli bebekler hariç, sekonder yayılım genellikle büyük primer tümör ile alakalıdır ve hastalığın geç döneminde oluşabilir.

56
Nöroblastoma Histopatoloji:
Makroskopik olarak yumuşaktır. Nekrotik ve hemorajik alanlar vardır Nörofibriler çekirdek etrafında nöroblastların oluşturduğu bir halka olan “Rozet Formasyonu” karekteristik bir bulgudur. Ancak her olguda görülmez. Histopatolojik olarak SHIMADA sınıflandırmasında; stromanın varlığı, NB hücresinin diferansiyasyon derecesi ve nüklear morfoloji (mitozis ve karyoreksis) ortaya konulur. Mitozis-karyorrhexis indeksi (MKI) hesaplanır (=her 5000 hücrede mitotik veya karyorektik hücrelerin sayımı) <100 hücre/ düşük orta > yüksek index. Buna göre iki prognostik gruba ayrılır: İyi ve kötü prognoz
 ortaya konulur. Mitozis-karyorrhexis indeksi (MKI) hesaplanır (=her 5000 hücrede mitotik veya karyorektik hücrelerin sayımı) <100 hücre/ düşük orta. > yüksek index. Buna göre iki prognostik gruba ayrılır: İyi ve kötü prognoz.")
57
Nöroblastoma Tümörün Yeri: Adrenal medullada %50-60
Retroperitonealde %20 Mediasteniumda %10 Pelviste %2-6 Boyunda %2 Genellikle ilk başvuru anında %62-70 oranında çocuklarda metastatik hastalık ardır. Lokalize hastalık %25 iken, Stage4S %10 civarındadır.

58
Tümör Markerları: Nöroblastoma
Nöroblastomaların %85 den fazlası yüksek katokolamin metabolitleri salgılarlar. En sık vanillylmandelik acid (VMA) ve homovanillic acid (HVA) ölçülür. VMA adrenalin ve nonadrenalin yıkım ürünüdür. HVA ise dopa ve dopamin’in metabolitleridir. Bu iki metabolitin idrar ve kandaki ölçümleri ile NB taraması ve tanısı mümkündür.
 ve homovanillic acid (HVA) ölçülür. VMA adrenalin ve nonadrenalin yıkım ürünüdür. HVA ise dopa ve dopamin’in metabolitleridir. Bu iki metabolitin idrar ve kandaki ölçümleri ile NB taraması ve tanısı mümkündür.")
59
Nöroblastoma Tümör Markerları: Biyokimyasal:
Nöron Spesifik Enolaz: Santral ve periferik sinir sistemi hücreleri ve nöroblastom hücreleri tarafından ekskrete edilen bir glikolitik enzimdir. 15 ng/ml den yüksek ise anormaldir. 100ng/ml den daha yüksek düzeyler ileri evreleri ve düşük sürvey oranları ile koreledir. Laktik Dehidrogenaz: 1500 IU/ml den az değerler iyi sürvey oranına işarettir. Ferritin: Nöroblastom hücreleri ferritin üretirler. Düşük evrelerde yüksek ferritin düzeyleri olağan değildir. 142 ng/ml üstü düzeyler Evre 3 ve 4 de görülür ve daha kötü gidişatı göaterir.

60
Nöroblastoma N-myc proto-onkogen,
Moleküler: N-myc proto-onkogen, diploid DNA kontenti (Hiperdiploidi), kromozom 1p’ nin delesyonu, CD44 ekspresyonu, Trk-A, MRP geni veya proteini
, kromozom 1p’ nin delesyonu, CD44 ekspresyonu, Trk-A, MRP geni veya proteini.")
61
Nöroblastoma Evreleme: Birden fazla evreleme şekli vardır.
Nöroblastoma-Pediatrik Onkoloji Grubu Evreleme Sistemi Evre A: Gross olarak komplet rezeksiyonu mümkün olan tümör, mikroskopik residü var veya yok. Evre B: Gros olarak rezeke edilemeyen tümör Evre C: Primer tümör komlet veya inkomplet rezeke edilebilir, ama primere yapışık olmayan intrakaviter nodlullerde histolojik olarak tümör pozitifliği. Evre D: İntrakaviter nodüllerin de ötesine disseminasyon vardır (ekstrakaviter nodlar, karaciğer,deri,kemik iliği, kemik gibi) Evre D(S): Evre 1 ve 2 olan tümörün karaciğer infiltrasyonu veya kemik iliğine metastazı (S=spesiyal)
 Evre D(S): Evre 1 ve 2 olan tümörün karaciğer infiltrasyonu veya kemik iliğine metastazı (S=spesiyal)")
62
Nöroblastoma Klinik: Karında kitle veya metastaz varlığına ait semptom ve bulgular: Bulantı, kilo kaybı, ateş, ve terleme genel semptomlar Kemik ve eklem ağrısı metastazın bir sonucu olup ilk başta juvenil artrit gibi tanı konulabilir Periorbital ekimoz veya proptozis ilk belirti olabilir Tümör boyunda ise kitle dışarıdan görülebilir Apikal toraks tümörlerinde Horner sendromu görülebilir Pelvik tümörlerde barsak ve mesane fonksiyonlarında değişiklik ve sfinkter sorunları görülebilir.

63
Nöroblastoma Klinik: Abdominal tümörlerin çoğunluğu semptomatik metastatik hastalık sonucu teşhis edilirler. Lokomotor rahatsızlıklar ve progresif parapleji de bir kısım nöroblastomalarda görülebilir. Fizik incelemede Abdominal nöroblastomlar genellikle sert, irregüler, ve fiksedir. Bazen bebeklerde yumuşak ve ballotable olabilir. Pelvik tümörler abdominal muayenede veya rektal muayenede ele gelebilirler.

64
Nöroblastoma Klinik: Evre IVS bir bebekte multipl deri lezyonları görülebilir. Sıklıkla hepatomegaliden dolayı abdominal distansiyon vardır. Hipertansiyon, yeni tanısı konulmuş NB’lularda %19 oranında görülür ve genellikle katekolamin sekresyonuna veya renal arter kompresyonuna bağlıdır. Kan basıncı ile üriner katekolamin sekresyonu arasında bir korelasyon bulunamamıştır. Hipertansiyon, tümör rezeksiyonu veya kemoterapi ile düzelir.

65
Nöroblastoma Klinik: Paraneoplastik sendromlar
Opsomyoclonus (dans eden gözler) sendromu; Progresif serebellar ataksi ile karekterizedir. Bu hastaların yarıdan fazlasında torakal primer tümör vardır.. Bunlar nöroblastoma göre ganglonöroma ve ganglionöroblastomada daha sıktır. İnatçı diareler ise nadirdir ve tümörden salgılanan vasoaktif intestinal polipeptide bağlıdır
 sendromu; Progresif serebellar ataksi ile karekterizedir. Bu hastaların yarıdan fazlasında torakal primer tümör vardır.. Bunlar nöroblastoma göre ganglonöroma ve ganglionöroblastomada daha sıktır. İnatçı diareler ise nadirdir ve tümörden salgılanan vasoaktif intestinal polipeptide bağlıdır.")
66
Nöroblastom

67
Rakun gözü bulgusu Masif hepatomegali (EvreIV S)
")
68
Nöroblastoma Tanı: Klinik bulgular sonucu şüphelenilen durumun konfirme edilmesi için laboratuvar ve rayolojik incelemelerin yanı sıra doku incelemesinin yapılması gerekir. Daha sonra evreleme araştırmaları ile tamamlanır. Labaratuvar araştırmaları: Üriner katekolamin metabolitlerinin ölçümü ilk tanısal taramadır. Üriner VMA ve HVA nöroblastomlu çoğu olguda yüksektir. Ayrıca tam kan sayımı, serum biyokimyası, karaciğer ve böbrek fonksiyon testleri, serum ferritin, LDH ve NSE ölçülmelidir.

69
Nöroblastoma Tanı: Görüntüleme:
Sırasıyla düz grafiler (ADKG, iki yönlü akciğer grafisi, kemik grafileri) USG, kontrastlı CT veya MRI ve MIBG sintigrafisi yapılır. Düz grafide; %50 den fazla distrofik kalsifikasyonlar görülür. Toraks grafisinde mediastinal tümör olup olmadığı görülebilir. USG ile kistik veya solid kitle ayrımı yapılır. Nöroblastomanın US grafik olarak diagnostik bir görüntüsü yoktur. Genellikle kalsifikasyon ve nekroz alanlarına sahip mixed eko paterni görülür. Pelvis ve toraksta USG kısıtlı kullanıma sahiptir.
 USG, kontrastlı CT veya MRI ve MIBG sintigrafisi yapılır. Düz grafide; %50 den fazla distrofik kalsifikasyonlar görülür. Toraks grafisinde mediastinal tümör olup olmadığı görülebilir. USG ile kistik veya solid kitle ayrımı yapılır. Nöroblastomanın US grafik olarak diagnostik bir görüntüsü yoktur. Genellikle kalsifikasyon ve nekroz alanlarına sahip mixed eko paterni görülür. Pelvis ve toraksta USG kısıtlı kullanıma sahiptir.")
70
Doğru bir anatomik detay için kontrastlı CT veya MRI gerekir. MRI
Görüntüleme: Doğru bir anatomik detay için kontrastlı CT veya MRI gerekir. MRI yumuşak doku değişiklikleri, karaciğer tutulumu ve intraspinal uzanım anatomisi (dumbbell tümörü) yönlerinden daha deyaylı bilgiler verir. Myelografiden daha üstün olup halihazırda kemik tutulumunun olup olmadığıda görülebilir. İskelet tutulumu düz grafilerde periostal erezyon, radiolüsensi ve patolojik kırıklar şeklinde kendini gösterebilir. Ancak kemik ve kemik iliği tutulumunu en iyi radioizotop işaretli “metaiodobenzylguanidine (MIBG)” sintigrafisi ortaya koyar. MIBG ile yalancı negatif sonuçlar elde edilebilir. Bu yüzden Tc99m metilen difosfonat (MDP) kemik sintigrafisi MIBG nin negatif olduğu hallerde kullanılır.
 yönlerinden daha deyaylı bilgiler verir. Myelografiden daha üstün olup halihazırda kemik tutulumunun olup olmadığıda görülebilir. İskelet tutulumu düz grafilerde periostal erezyon, radiolüsensi ve patolojik kırıklar şeklinde kendini gösterebilir. Ancak kemik ve kemik iliği tutulumunu en iyi radioizotop işaretli metaiodobenzylguanidine (MIBG) sintigrafisi ortaya koyar. MIBG ile yalancı negatif sonuçlar elde edilebilir. Bu yüzden Tc99m metilen difosfonat (MDP) kemik sintigrafisi MIBG nin negatif olduğu hallerde kullanılır.")
71
Nöroblastom

72
Nöroblastom MIBG sintigrafisi

75
Tümör

79
Nöroblastoma Tanı: Doku biyopsisi: Tanıda doku biyopsisi esastır.
Kemik iliği aspirasyonu, primer veya sekonder hastalığın biopsisi ile doku alınabilir. Tanı ve sitogenetik çalışmalar için yeterli doku alınmalıdır.

80
Tedavi: Nöroblastoma Nöroblastomun tedavisi multidisiplinerdir.
Tedavi; cerrahi, kemoterapi ve radyoterapi içerir. Cerrahın rolü; mümkün olduğunca tümörün çıkarılması, gerekirse biyopsi alınması ve vasküler yolun sağlanmasıdır.

81
Nöroblastoma Prognoz 1970’li yıllarda 5 yıllık sürvi oranı %15 iken günümüzde oldukça iyi bir duruma gelmiştir. 1980 –1990 yılları arasında tüm sürvi oranı %58 olarak hesaplanmıştır. Prognozu etkileyen birçok faktörler vardır.

82
Prognostik faktörler Nöroblastoma
Ganglonöroma’ya maturasyonu: Nöroblastoma spontan olarak regrese olabilen tek tümördür. Bunun insidansı bilinmiyor. Çok yeni taramalar göstermiştir ki; tedavi edilmemiş tümörlerde spontan regresyon olmuştur.

83
Prognostik faktörler Erken tanı ve tarama Nöroblastoma
Antenatal olarak saptanan tümörler favorable biyolojik profil gösterir. Tarama sonucu bulunan tümörler “favorable” olup ve sürvi şansı %92’dir. Bu grupta spontan regresyon sıktır.

84
Prognostik faktörler Nöroblastoma Biyokimyasal ve Moleküler Marker:
Bahsedilen çeşitli marker varlığı veya düzeyleri tümörün akıbetini etkiler.

85
Prognostik faktörler Nöroblastoma Başvuru yaşı:
1 yaş altında tanı almış çocuklarda sürvi oranı %84 iken 1 yaş üstündekilerde %42 dir.

86
Prognostik faktörler Paraneoplastik sendromlar Nöroblastoma
Paraneoplastik sendromların görüldüğü hastalarda mortalite oranı sürpriz bir şekilde düşüktür.

87
Prognostik faktörler Nöroblastoma Orijin aldığı yer
1970’lerde sürvi oranları servikal ve pelvik primer tümörlerde %100, torasiklerde %75 ve abdominal tümörlerde %28 di. 1990’ların ortalarında torasiklerde %83 olarak bildirilmiştir. Servikal ve torasik primer tümörü olan bebeklerin çoğunluğunda hiperdiploid DNA kontenti görülür. Spinal kanala uzanımı Dumbbell tümörlerinde yüksek sürvi oranıları söz konusudur.

88
Prognostik faktörler Nöroblastoma Evreleme ve Stage IVS
Düşük evrelerde sürvi daha yüksektir. Evre IVS hastalarda sürvi %50 civarındadır. Adjuvan terapi: Lokal olarak yayılmış veya metastatik hastalıklar için, myeloablative tedavi ile kombine multiajan kemoterapi sürvi oranını artırmıştır.

89
Nöroblastoma-Prognozu etkileyen faktörler
İyi Prognoz Kötü Prognoz Yaş <1 yaş >1 yaş Evre I, II, IV-S III, IV Shimada histoloji Stroma zengin Stroma fakir Yeri Mediatenum, pelvis, boyun Adrenal, çölyak aks >10 N-myc kopyası Hayır Evet DNA-flow sitometri Hiperdiploid Diploid Yükselmiş trk-A Yüksek serum Ferritin Nöron-spesifik enolaz Laktat dehidrogenaz Kr. 1 heterozigosite yokluğu Multidrug rezistant-gen Multidrug rezistant protein-gen Somatostatin reseptörleri Vazoaktif intestinal peptid sekresyonu MHC class 1 antijeni Tarama sonucu saptanma

90
Karaciğer Tümörleri

91
Karaciğer Tümörleri Çocukluk çağında hepatik orijinli tümörler sık olmamasına karşın tüm karaciğer lezyonlarının yaklaşık ¾ ü malign’dir. Pediatrik hepatik kanserlerin %90’dan fazlasını Hepatoblastoma (HB) ve Hepatocellular carsinoma (HCC) oluşturur. Primer hepatik malignansilerin %65’ini HB, %25’ini HCC oluşturur
 ve Hepatocellular carsinoma (HCC) oluşturur. Primer hepatik malignansilerin %65’ini HB, %25’ini HCC oluşturur.")
92
Çocukluk çağı primer hepatik tümör insidansı
Hepotoblastoma 43 Hepatoselüler Ca 23 Sarkom 6 Benign vasküler tümör 13 Mezenşimal hamartom Adenoma 2 Fokal nodüler hiperplazi Diğerleri 5

93
Karaciğer Tümörleri HB ve HCC; insidans oranları,
epidemiyolojik özellikler, tedaviye cevap ve prognoz yönleriyle birbirinden oldukça farklıdırlar.

94
Karaciğer Tümörleri EPİDEMİYOLOJİ
HB; tipik olarak 1-3 yaş çocuklarda gelişen bir embriyonel tümördür. Olguların çoğunluğu sporadiktir. Ancak bazı spesifik sitogenetik anormalliklerde rölatif artış olabilir. En sık kromozom 2 ve 20’nin trizomisi ve kromozom 11’in kısa kolunda heterozigosity yokluğu dikkati çeker.

95
Karaciğer Tümörleri EPİDEMİYOLOJİ
HB; Beckwith-Wiedeman sendromlu (BWS) çocuklarda yüksek sıklıkta görülür. Aynı zamanda hemihipertrofili, renal ya da adrenal agenezili kişilerde ve familial adenomatöz polipozisili olguların çocuklarında sıklığı artar. BWS geninin yerleştiği 11p15; tümör süpresor lokusunun varlığını gösterir.
 çocuklarda yüksek sıklıkta görülür. Aynı zamanda hemihipertrofili, renal ya da adrenal agenezili kişilerde ve familial adenomatöz polipozisili olguların çocuklarında sıklığı artar. BWS geninin yerleştiği 11p15; tümör süpresor lokusunun varlığını gösterir.")
96
Karaciğer Tümörleri EPİDEMİYOLOJİ
HCC; çocukluk çağında erişkindeki gibi agresif biyolojik davranış gösterir. Sıklıkla; daha önceden karaciğer hastalığı olan kişilerde ve hepatit B’nin endemik olduğu Afrika ve Asya’nın belli bölümlerinde görülür. Bu bölgelerde HCC insidansı 200 kez daha sıktır. Bu ülkelerde en sık görülen primer karaciğer tümörü HCC’dir. Hepatit B infeksiyonun yüksek olan popülasyonlarda hepatit B aşısı ile rutin immunizasyon bir efektif kanser kontrol stratejisidir.

97
Karaciğer Tümörleri EPİDEMİYOLOJİ
HCC; metabolik ve inflamatuvar hepatopatilerin diğer tiplerine sahip hastalardada gelişebilir. Tirozinozis, glikojen depo hastalığı, alfa-1 antitripsin eksikliği, neonatal hepatit ve bilier atrezi gibi durumlar HCC gelişimi için yüksek risk oluştururlar.

98
Karaciğer Tümörleri KLİNİK
HB; tipik olarak ilk 3 yaş sırasında (ort:3,5 yaş) görülür HCC’li olguların çoğunluğu okul çağı çocuğudur. (ort 11,2 yaş). Her iki tümör de erkek çocuklarda fazla görülmektedir (erkek/kız oranı 3/1 dir). Abdominal kitle %68, abdominal distansiyon %28, anoreksi %23, kilo kaybı %23 abdominal ağrı %19 ve kusma %11 görülür. Ateş ve trombositozis sıklıkla gözlenir.
 görülür. HCC’li olguların çoğunluğu okul çağı çocuğudur. (ort 11,2 yaş). Her iki tümör de erkek çocuklarda fazla görülmektedir (erkek/kız oranı 3/1 dir). Abdominal kitle %68, abdominal distansiyon %28, anoreksi %23, kilo kaybı %23 abdominal ağrı %19 ve kusma %11 görülür. Ateş ve trombositozis sıklıkla gözlenir.")
99
Karaciğer Tümörleri TANISAL GÖRÜNTÜLEME
Hem preoperatif tanıda hem de postoperatif izlemde radyolojik tanısal görüntüleme önemlidir. Diagnostik görüntülemede amaç; benign-malign ayırımı, tanının belirlenmes ve rezeke edilebilirliğin belirlenebilmesidir. Bu amaçla ; standart abdominal ve toraks radiografileri alınır. Daha sonra abdominal ultrasonografi (US) önerilir.
 önerilir.")
100
Karaciğer Tümörleri TANISAL GÖRÜNTÜLEME
US; kitlenin sınırları, uzanımı (solit/multifokal), komşulukları, natürü (kistik/solid veya karışık), kanlanması ve kan damarları hakkında yararlı bilgiler sağlar. US hatta perkütan biopsi amacıyla kullanılabilir. Bazen da intraoperatif olarak tümörün uzanımını belirlemede kullanılabilir.
, komşulukları, natürü (kistik/solid veya karışık), kanlanması ve kan damarları hakkında yararlı bilgiler sağlar. US hatta perkütan biopsi amacıyla kullanılabilir. Bazen da intraoperatif olarak tümörün uzanımını belirlemede kullanılabilir.")
101
Karaciğer Tümörleri TANISAL GÖRÜNTÜLEME
CT ve MRI incelemeleri de mutlaka yapılması gerekir. MRI; Tümörün hepatik vasküleriteye komşuluklarını tam belirlemede CT’den üstünlükleri vardır. Her hastaya preoperatif olarak, metastatik hastalık açısından toraks CT’si çekilir. Pulmoner metastazlı çocuklar ve ekstensif bilobar veya multifokal tümörlü olgular properatif kemoterapi adayıdırlar. Majör karaciğer rezeksiyonunda komplikasyon sıktır.

102
Karaciğer Tümörleri TANISAL GÖRÜNTÜLEME
MRI ve spiral CT’nin ilerlemesi ile, arteriografi çocukluk çağı hepatik tümörlülerde tanısal amaçlı nadir kullanılmaktadır. Arteriografi sıklıkla terapötik amaçlı (kemoembolizasyon ve sitotoksik ilaçların intraarterial infüzyonu) kullanılır. Günümüzde diagnostik arteriografi belki inkomplet rezeksiyon yapılmış ve reoperasyon planlanan olgular için rezerve edilmelidir.
 kullanılır. Günümüzde diagnostik arteriografi belki inkomplet rezeksiyon yapılmış ve reoperasyon planlanan olgular için rezerve edilmelidir.")
103
Karaciğer Tümörleri LABORATUVAR ÇALIŞMALARI
CBC, trombosit sayımı, koagülasyon profili, ve karaciğer fonksiyon testleri çalışılmalıdır. Anemi sık görülür. Trombositozis, hastaların %60’ı oranında olduğu bildirilmiştir.

104
Karaciğer Tümörleri LABORATUVAR ÇALIŞMALARI
Hem tanısal hem de terapötik bakımdan en informatif laboratuar testi; serum alfa-fetoprotein (AFP) çalışmasıdır. AFP, İlk prezantasyon anında, hepatik epiteliyal malignansili çocuklarda %93’ündeyükselmiştir. AFP düzeyleri tümör aktivitesi ile iyi korele olmasına ve hepatik epiteliyal malignansi varlığını göstermesine karşın, artmış AFP, HB ve HCC için kesin tanısal değildir. Artmış AFP düzeyleri; çocuklarda yolk sac neoplazmları, mezenkimal hamartomlar, sarkomlar ve hemangioendoteliomalar’da da söz konusudur.
 çalışmasıdır. AFP, İlk prezantasyon anında, hepatik epiteliyal malignansili çocuklarda %93’ündeyükselmiştir. AFP düzeyleri tümör aktivitesi ile iyi korele olmasına ve hepatik epiteliyal malignansi varlığını göstermesine karşın, artmış AFP, HB ve HCC için kesin tanısal değildir. Artmış AFP düzeyleri; çocuklarda yolk sac neoplazmları, mezenkimal hamartomlar, sarkomlar ve hemangioendoteliomalar’da da söz konusudur.")
105
Karaciğer Tümörleri LABORATUVAR ÇALIŞMALARI
HCC’de ilk AFP ölçümünün büyüklüğü tanısal öneme sahip olabilir. HCC’de yüksek AFP kötü prognozu gösterir. HB’de ise bunun tersini gösteren çalışmalar vardır. Ancak follow-up yönünden AFP’nin önemi tartışılmaz. AFP’nin yarı ömrü 4-8 gündür. Rezidü tümör yoksa linear tarzda bir düşüş gösterir.

106
Karaciğer Tümörleri EVRELEME
TANIM I Primer hepatik tümörün komplet rezeksiyonu. Bilinen residü hastalık yok. Favorable / unfavorable histoloji II Total gross rezeksiyon, mikroskopik residü hastalık bulgusu var İntrahepatik / ekstrahepatik III Cerrahi sonrası gross residual tümör var. Komplet rezeke edilen primer hepatik tümör fakat nodüller pozitif veya tümör spill sözkonusu. IV Metastatik hastalık Komplet rezeke edilen primer hepatik tümör Komplet rezeke edilemeyen primer hepatik tümör

107
Karaciğer Tümörleri PATOLOJİ
Hepatoblastomanın histolojik subtipleri: Epitelial Fetal Embryonal Macrotrabeküler Küçük hücre Mixed Epitelial / Mezenkimal Teratoid Nonteratoid

108
Karaciğer Tümörleri TEDAVİ
Günümüzdeki tedavi stratejileri; cerrahi, kemoterapi ve transplantasyon kombinasyonlarıdır ve bu strateji tümörün PRETEXT evresi ile belirlenir. PRETEXT: Preoperatif evreleme sistemidir (SIOP). Burada hastalığın evresini tutulan karaciğer sektör (Couinaud segmentleri) sayısı belirlemektedir. Cerrahi rezeksiyon ve kemoterapi gerekir. İlk laparotomide total rezeksiyon çok önemlidir. Rezeke edilemeyen tümör söz konusu ise preoperatif kemoterapi ile tedaviye başlanır ve geç hepatik rezeksiyon yapılır. HB’li çocuklarda prognoz HCC’den çok iyidir. İlk rezeksiyon HCC’de daha hayati önem taşır. Çünkü HCC’nin kemoterapiye yanıtı çok kötüdür.
. Burada hastalığın evresini tutulan karaciğer sektör (Couinaud segmentleri) sayısı belirlemektedir. Cerrahi rezeksiyon ve kemoterapi gerekir. İlk laparotomide total rezeksiyon çok önemlidir. Rezeke edilemeyen tümör söz konusu ise preoperatif kemoterapi ile tedaviye başlanır ve geç hepatik rezeksiyon yapılır. HB’li çocuklarda prognoz HCC’den çok iyidir. İlk rezeksiyon HCC’de daha hayati önem taşır. Çünkü HCC’nin kemoterapiye yanıtı çok kötüdür.")
109
Karaciğer Tümörleri TEDAVİ
TRANSPLANTASYON Ekstrahepatik tutulumu olmayan ve kemoterapi sonrası komplet rezeke edilemeyen olgularda önerilmektedir KEMOTERAPİ Tüm HB ve HCC’li olgulara kemoterapi (rezeksiyon sonrası ve / veya rezeksiyon öncesi) gerekir. RADYOTERAPİ HB ve HCC’de radyoterapinin sınırlı bir kullanımı söz konusudur. HB radyosensitif bir tümördür, ama HCC’de radyoterapi etkili değildir.
 gerekir. RADYOTERAPİ. HB ve HCC’de radyoterapinin sınırlı bir kullanımı söz konusudur. HB radyosensitif bir tümördür, ama HCC’de radyoterapi etkili değildir.")
110
Karaciğer Tümörler SONUÇ
Geçtiğimiz 20 yıl süresince HB’li çocukların prognozunda oldukça iyi bir ilerleme olmasına karşın HCC’de çok küçük değişiklikler olmuştur. Bu tümörlerin düşük insidansları dolayısıyla çok merkezli pediatrik kanser çalışma gruplarına ihtiyaç vardır.

111
Teratomlar

112
Teratomlar EMBRİYOLOJİ ve TERATOMA
Eski yunanca’da teratos (dev)ve onkoma (şişlik) kelimelerinden köken alır. Totipotent primordial germ hücrelerinden gelişir. Her ne kadar teratomlar embriyonik üç tabakadan (endodermal, mezodermal, ektodermal tabakaları) köken alan tümörler olarak tanımlansa da günümüzde monodermal tipleri de içeren sınıflandırmalar yapılmaktadır.
ve onkoma (şişlik) kelimelerinden köken alır. Totipotent primordial germ hücrelerinden gelişir. Her ne kadar teratomlar embriyonik üç tabakadan (endodermal, mezodermal, ektodermal tabakaları) köken alan tümörler olarak tanımlansa da günümüzde monodermal tipleri de içeren sınıflandırmalar yapılmaktadır.")
113
Teratomlar EMBRİYOLOJİ ve TERATOMA
Birçok tümör deri elementleri, nöral dokular, diş, yağ, kıkırdak, normal ganglion hücreli intestinal mukoza içerirler. Bu dokular genellikle disorganize şekilde yerleşirler. Bazen oldukça organize dokular içeren (ince barsak, ekstremite ve hatta çalışan kalp) tümöre fetiform teratom adı verilir. Eğer kitle vertebra veya notokord içeriyor ve yüksek derecede organize yapı varsa buna fetus in fetu adı verilir. Doğumda malignansi çok nadirdir, ancak yaş ve inkomplet rezeksiyon ile bu risk artar.
 tümöre fetiform teratom adı verilir. Eğer kitle vertebra veya notokord içeriyor ve yüksek derecede organize yapı varsa buna fetus in fetu adı verilir. Doğumda malignansi çok nadirdir, ancak yaş ve inkomplet rezeksiyon ile bu risk artar.")
114
Teratomlar Teratomlar genellikle izole lezyonlardır. Anorektal malformasyon, sakral anomali ve presakral kitle (genellikle teratom veya meningosel’ dir, nadiren duplikasyon kisti veya dermoid kisti olabilir) triadı çok iyi ortaya konmuştur.
 triadı çok iyi ortaya konmuştur.")
115
Teratomlar BİYOLOJİK MARKERLAR
ALFA-FETO PROTEİN: Bir alfa-globülindir. AFP’nin esas kaynağı fötal karaciğerdir. Endodermal sinüs tümörlerinde ve embriyonel karsinomda AFP yüksektir. AFP düzeylerinin izlenmesi rezeksiyonun yeterliliği açısından önem taşır. Aynı zamanda erken tümör rekürrensinin saptanmasında faydalıdır. AFP başka hastalıklarda da yükselebilir: Hepatik malignensiler, hipotiroidizm, atexia telengectasia, hetit, gastrointestinal malignensiler, ve herediter tirozinemi. Tüm bunların germ hücreli tümörlerden ayırt edilmesi gerekir.

116
Teratomlar BİYOLOJİK MARKERLAR
BETA HUMAN KORİONİK GONADOTROPİN (β-hCG): Trofoblastik elementler içeren germ hücreli tümörlerde (koriokarsinom) saptanır. Ayrıca bazı testiküler tümörlerde, hepatomada, hepatoblastomda, pineal bezin germinomunda beta-HCG saptanabilir. Yarılanma ömrü 45 dakika olduğundan, tümör rezeksiyonundan hemen sonra hızlıca kaybolması gerekir. Nadiren de karsinoembriyonik antijen (CEA) bir marker olabilir.
: Trofoblastik elementler içeren germ hücreli tümörlerde (koriokarsinom) saptanır. Ayrıca bazı testiküler tümörlerde, hepatomada, hepatoblastomda, pineal bezin germinomunda beta-HCG saptanabilir. Yarılanma ömrü 45 dakika olduğundan, tümör rezeksiyonundan hemen sonra hızlıca kaybolması gerekir. Nadiren de karsinoembriyonik antijen (CEA) bir marker olabilir.")
117
Teratomlar Lokalizasyon
Sakrokoksigeal 47,2 Over 27,1 Beyin / CNS 3,6 Göğüs 5,1 Farenks 2,2 Servikal 4,5 Pelvik 0,8 Retroperitoneal 4,0 Diğer 1,0

118
Teratomlar KLİNİK Çoğunlukla sakrokoksigeal teratomlar doğumda gözle görülür şekildedir. Kızlarda 3 kat fazla görülür. Ayırıcı tanıda meningosel ve lenfanjiom göz önünde bulundurulmalıdır. Radyolojik olarak düz grafiler, USG ve MRI kullanılarak, tümörün büyüklüğü, uzanımı ve komponenti hakkında bilgi edinilir. Tedavide esas tümörün komplet çıkarılmasıdır. İnkomplet rezeksiyonlar ile malignansi riski artar.

119
Teratomlar KLİNİK Prenatal USG ile tanısı konulabilir. Doğumda ise kitle 5 cmden veya fötüs çapından büyük ise sezeryan önerilmektedir. Distosi sonucu tümör rüptürü ve kanama olabilir. Sakrokoksigeal teratomlar fötal cerrahi uygulaması yapılan ender patolojilerden biridir. Sakrokoksigeal teratomların sonuçları genellikle iyidir.

120
Teratomlar KLİNİK Torasik teratomlar: Mediastinal teratomlar ve Intraperikardiyal teratomlar Abdominal teratomlar: Retroperitoneal teratomlar ve Gastrik teratomlar Baş ve Boyun Teratomları: Servikal teratomlar, Kraniofasiyal teratomlar (Epignatus, Pharengeal teratomlar, Orofarengeal teratomlar, Orbital teratomlar, Orta kulak teratomları) ve İntrakraniyal teratomlar Diğerleri: Deri, koksiks’den uzak perianal bölge, umbilikal kord ve plasenta gibi diğer yerlerde de teratomlar bidirilmiştir.
 ve İntrakraniyal teratomlar. Diğerleri: Deri, koksiks’den uzak perianal bölge, umbilikal kord ve plasenta gibi diğer yerlerde de teratomlar bidirilmiştir.")
121
Germ Hücre Tümörlerinin Klasifikasyonu (embryolojik yakınlık temelinde)
Germinoma Disgerminoma Seminoma Embryonik diferansiyasyon (embryonel karsinoma) Somatik diferansiyasyon Matür teratoma Immatüre teratoma Malign teratoma Ekstraembryonik diferansiyasyon Yolk sac endodermal sinüs tümörü Trofoblast koryokarsinoma
 Somatik diferansiyasyon. Matür teratoma. Immatüre teratoma. Malign teratoma. Ekstraembryonik diferansiyasyon. Yolk sac endodermal sinüs tümörü. Trofoblast koryokarsinoma.")
122
Germ Hücre Tümörlerinin Karşılaştırmalı Klinik Prezentasyonları
YAŞ % SEMPTOMLAR BULGULAR PATOLOJİ Ekstragonadal Sakrokoksigeal Bebek 41 Konstipasyon, mesane veya alt ekstremitede nörolojik anormallikler Presakral kitle %65 benign, %5 immatür %30 malign Mediastinal 2 yaş altı 6 Öksürük, weezing, dispne Anterior mediastinal kitle Benign veya malign Abdominal 5 Bası ağrısına sekonder, genitoüriner obst Sıklıkla retroperitoneal İntrakranial Çocukluk Baş ağrısı, inkoordinasyon Pineal veya supraselüler tümörler; serebrospinal sıvıda AFP veya HCG Herhangi tip germ hücreli tümör Baş ve boyun 4 Bası ile ilgili; solunum veya yutma güçlüğü Büyük kitle Genellikle benign Vajina 3 yaş altı 1 Kan boyalı vajinal akıntı Vajinadan polipoid kitle Genellikle malign Gonadal Ovarian 10-14 yaş 29 Abdominal ağrı, bulantı, kusma, konstipasyon, genitoüriner semptomlar Abdominal pelvik kitle, %50sinde kalsifikasyon, sıklıkla AFP veya HCG; gebeliği taklit edebilir Testiküler Bebek, post- pubertal 7 Testisi ağrısız şişliği, ağrılı torsiyon Testiküler kitle; bebeklerde akciğere metastaz Herhangi tip germ hücreli tümör; %82 malign, %18 benign; bebekte çoğunlukla yolk sac tümörler

123
Germ Hücre Tümörleri TEDAVİ
Germ hücreli tümörlerin tedavisi; polimorf histoloji, multipl lokasyonlar, klinik evre ve hastanın yaşına göre değişir. Tedavide; ilk “debulking” cerrahi, yoğun multimodal kemoterapi ve bazen radyasyon tedavisi gibi değişik kombinasyonlar kullanılır. Temel olarak tüm germ hücreli tümörler komplet cerrahi rezeksiyonla tedavi edilirler. Benign germ hücreli tümörlerde tek başına cerrahi ile kür sağlanır. Rezektabiliteyi belirleyen en önemli faktör anatomik lokalizasyondur. İmmatür elementler içeren tümörler geniştir ve rezeksiyonları zordur.

124
Germ Hücre Tümörleri PROGNOZ
1970 lerde %10 olan tüm yaşam oranı günümüzde %90’ların üstündedir. Malign germ hücreli tümörlerde kötü prognoz faktörleri: ekstragonadal yerleşim, 11 yaşından büyük olma, hastalığın uzanımı, komplet rezeksiyon yapılamaması ve germinom veya miks germ hücreli tümör histolojisi.

Benzer bir sunumlar



>")

>")















