Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
HEMOLİTİK ANEMİLER Dr. Ali ünal

2
Normal Periferik Yayma

3
ERİTROPOEZ Eritrositlerin kemik iliğinde en erken hücresi proeritroblasttır. Proeritroblast bölünme sürecine girerek sırasıyla bazofilik eritroblast, polikromatofilik eritroblast, ortokromik eritroblast evrelerinden geçerek sonuçta retikülosit ve eritrosit oluşur.

5
Eritrosit turnoveri Proeritroblast döneminden retikülosit oluşana kadar geçen süre 5 gündür. Perifere çıkan retikülositler saat içerisinde olgun eritrosit şeklini alırlar. Eritrositlerin yaşam süresi ise ortalama 120 gündür. Normal Hemostatik şartlarda yapılan eritrosit kadarı yıkılmaktadır. Erişkin bir kişide günde 5x104/ul eritrosit yapılmaktadır Normal koşullrda yaşlanan eritrositler karaciğer ve dalakta RES fagositik hücreleri tarafından uzaklaştırılarak yok edilir.

7
Hemoliz (yıkım) YERİ….. İntravasküler hemolizde eritrositler dolaşımda parçalanır ve eritrosit içeriği plazmaya karışır. Daha sık görülen ekstravasküler hemolizde ise eritrositler makrofajlar tarafından KC ve dalak ve RES te fagositoza ugrar.

8
İntravasküler hemoliz;
Eritrositlerin travmayla karşılaşması (protez kapak), Eritrositlere kompleman bağlanması (immün hemolitik anemi) Ortamda bulunan ekzojen toksik faktörlerin (bakteri toksini) saldırısına uğraması sonucu oluşur
, Eritrositlere kompleman bağlanması (immün hemolitik anemi) Ortamda bulunan ekzojen toksik faktörlerin (bakteri toksini) saldırısına uğraması sonucu oluşur.")
9
İntravasküler hemolizde; hemoglobin
Eritrositler periferik dolaşımda iken hemolize uğradığı için eritrosit içinde bulunan maddeler plazmaya geçer. Hb’de bu plazmaya geçen maddelerdendir. Ciddi intravasküler hemolizde plazma Hb konsantrasyonu artar Hb, haptoglobuline bağlanır.. Haptoglobulin plazmadaki serbest Hb’ini bağlayıp Hb’ini vasküler yatağın dışına kaçırarak idrar ile idrah edilmesi engellenmiş olur.

10
İntravasküler hemolizde; haptoglobulin
Plazmada mg/dl Hb’ini bağlayabilecek kadar haptoglobulin bulunur. Hb-haptoglobulin kompleksi dakikalar içinde hepatosit ve RES tarafından plazmadan uzaklaştırılır. İntravsküler hemolizde haptoglobulin katabolizma hızı yapım hızını geçer, bu nedenle plazma haptoglobulin düzeyi ölçülemeyecek kadar düşer ya da kaybolur.

11
İntravasküler hemolizde; LDH
Eritrositlerin içerinde bol bulunan bir enzimdir. İntravasküler hemoliz durumunda LDH düzeyleri çok yükselebilir.

12
Parçalanan eritrositlerden açığa çıkan Hb RES hücreleri tarafından indirek Bilüribin’e çevrilir ve serum indirek Bilüribin düzeyi artar.

13
Ekstravasküler hemoliz;
Extravasküler hemoliz olabilmesi için eritrosit membranına bağlı antikorun olması veya eritrosit deformabilitesini etkileyen bazı fiziksel faktörlerin olması gerekir. Eritrositler esas olarak dalak ve/veya KC’de bulunan Makrofajlar’lar tarafından fagosite edilirler ve parçalanırlar

14
Ekstravasküler hemolizde de
Haptoglobulin aynı şekilde düşer, Ancak plazma Hb düzeylerinde belirgin artış olmadığı için hemoglobinüri ve hemosiderinüri beklenmez. Serum LDH düzeyleri intravasküler hemolizdeki değerlerden daha az olmak üzere yine yüksektir. İndirek Bilüribin yine yüksek bulunur.

15
Hemoliz & retikülositoz
Ciddi hemoliz varsa Kemik İliğinde eritroid hiperplazi periferik kanda retikülositoz vardır.

16
Kalıtsal hemolitik anemilerde
Sarılık, Splenomegali, Hemolitik-aplastik krizler, Kolelitiazis, Bacak ülserleri Kemik anormallikleri olabilir.

17
Edinsel hemolitik anemiler
Akut olarak gelişirse klinik tablo çok ağırdır. Daha çok ateşli bir hastalığı anımsatır; sırt ağrısı, karın ağrısı, baş ağrısı, halsizlik, bulantı, kusma, titreme ile yükselen ateş ve şok-komaya kadar uzanan klinik spektrum olabilir. Solukluk, sarılık, taşikardi, terleme, çarpıntı ve anksiyöz bir durum izlenebilir. Yavaş gelişirse hastanın CVS’i anemiye adapte olacağından, klinik tablo konjenital anemilerde tarif edilene son derece benzer olabilir ve bu nedenle sadece deri ve skleralardaki sarılık olabilir

18
indirek hiperbilirubinemi tüm hemolizlerde izlenir.
Splenomegali ve indirek hiperbilirubinemi tüm hemolizlerde izlenir.

19
Eritropoez artış göstergeleri;
İntramedüller eritroid hiperplazi: (Kemik iliği: eritroid hiperplazi ) Ekstramedüller eritropoezis: Özellikle konjenital hemolitik anemilerde (H.sferositoz, talasemi, vs) Safra kesesi pigment taşları: Daha çok kalıtsal hemolitik anemilerde izlenir. Periferik kanda normoblast görülmesi:
 Ekstramedüller eritropoezis: Özellikle konjenital hemolitik anemilerde (H.sferositoz, talasemi, vs) Safra kesesi pigment taşları: Daha çok kalıtsal hemolitik anemilerde izlenir. Periferik kanda normoblast görülmesi:")
20
Retikülositoz: Kİ’nin hemolize efektif yanıtı periferk kanda bulunan retikülosit sayısı ile gösterilebilir. Kemik iliğinin kompansasyon derecesini daha sağlıklı olarak göstermesi bakımında daha çok düzeltilmiş retikülosit sayısı kullanılır. Düzeltilmiş retikülosit sayısı=Ret sayısı (%)X (hasta Htc/0.45)
X (hasta Htc/0.45)")
21
Hemolizde Laboratuar:
Retikülositoz LDH yükselir İndirekt biluribin yükselir Haptoglobin düşer Serumda methemalbuminemi ve serbest Hb bulunabilir İdrarda hemosiderinüri, Hbüri, methemalbuminüri olabilir Radyoaktif Cr işaretli eritrosit ömrü kısalabilir (Talasemi minör hariç). OEH ve OEHbK normal veya artmıştır.
. OEH ve OEHbK normal veya artmıştır.")
22
PY’da; Çekirdekli eritrositler, makrositler ve polikromazi → Hızlı hemoliz durumlarında görülebilir. Orak hücreler → Orak hücreli anemi Parçalanmış eritrositler(şistosit) → MAHA, prostetik kapak Akantosit(spur hücreler) → Abetalipoproteinemi Sferosit → Herediter Sferositoz Hedef hücre → Talasemi, Kc hast, Hb C Heinz cisimcikler → Unstabl Hb, G6PD eksikliğini düşündürür.
 → MAHA, prostetik kapak. Akantosit(spur hücreler) → Abetalipoproteinemi. Sferosit → Herediter Sferositoz. Hedef hücre → Talasemi, Kc hast, Hb C. Heinz cisimcikler → Unstabl Hb, G6PD eksikliğini düşündürür.")
23
EDİNİLMİŞ HEMOLİTİK ANEMİ SEBEPLERİ
Hipersplenizm İmmün hemolitik anemiler; OİHA, Alloimmün PNH (paroksismal noktürnal hemoglobinüri) Toksin ve ilaçlara bağlı hemoliz Mekanik travmatik (protez kapak) hemoliz Eritrosit parazitleri (sıtma) Spur hücreli anemi
 Toksin ve ilaçlara bağlı hemoliz. Mekanik travmatik (protez kapak) hemoliz. Eritrosit parazitleri (sıtma) Spur hücreli anemi.")
24
TRAVMATİK HEMOLİTİK ANEMİ
Travmatik kardiak protez kapak hemolitik anemisi March Hbürisi MAHA

25
MARCH HEMOGLOBİNÜRİSİ
Uzun yürüyüşler, koşu ve ağır sporlar tekrarlayan küçük damar travması sebebi ile IV hemolize yol açabilir. Hafif bir hemolizdir. Periferik yayma normaldir, tedavi gerekmez

26
FRAGMENTASYON HEMOLİZİ
Türbülan kan akımının eritrositleri zedelemesiyle oluşur. Aort stenozu ve yetmezliği, sinüs valsalva rüptürü, AV fistül, ve prostetik kapak (*en sık aort ve özellikle kapak disfonksiyonu varsa) hastalarında da travmatik hemoliz olabilir. Prostetik kapaklardaki hemoliz daha ciddidir.
 hastalarında da travmatik hemoliz olabilir. Prostetik kapaklardaki hemoliz daha ciddidir.")
27
MAHA (Mikroanjiopatik hemolitik anemi)
Hemoliz bulgularının yanı sıra ateş, kanama diatezi, trombositopeni, nörolojik bozukluklar ve böbrek yetmezliği vardır. Arteriol ve kapillerlerde hyalin mikro-trombuslar saptanır. Tedavide plazma değişimi (plazmaferez) en yararlı metottur. NEDENLERİ Dev hemanjiom (kasabach-meritt s), renal transplant reddi, Malign HT, eklampsi, PAN, wegener, Metastatik tümörler, TTP, HUS, E coli ve Mitomicin-C MAHA yapan sebepler arasında yer alır
 en yararlı metottur. NEDENLERİ. Dev hemanjiom (kasabach-meritt s), renal transplant reddi, Malign HT, eklampsi, PAN, wegener, Metastatik tümörler, TTP, HUS, E coli ve Mitomicin-C MAHA yapan sebepler arasında yer alır.")
28
TTP & HUS TTP: HÜS: 1. MAHA, 2. Trombositopeni, 3. Üremi,
4. Nörolojik bulgular, 5. Ateş HÜS: 2. Üremi, 3. Trombositopeni

29
MAHA (Mikroanjiopatik hemolitik anemi)
")
30
OTOİMMÜN HEMOLİTİK ANEMİLER (OİHA)
Hastanın kendi eritrosit antijenlerine karşı antikor üretmesi ve bunun sonucunda eritrositlerin yıkılmasıdır. 1- IgG 2- IgM tipinde iki grup antikor tarafından oluşturulabilir.

31
1. Sıcak antikor aracılıklı OİHA (IgG bağımlı)
**Bu tür, OİHA’nin en sık türü olup (%70-80), eritrositler dalakta sekestre olmaktadır. Hemoliz ekstravasküler alanda gerçekleşir. Hemolizin oluşması için kompleman aktivasyonuna gerek yoktur ama kompleman aktivasyonu hemolizi hızlandırır.
, eritrositler dalakta sekestre olmaktadır. Hemoliz ekstravasküler alanda gerçekleşir. Hemolizin oluşması için kompleman aktivasyonuna gerek yoktur ama kompleman aktivasyonu hemolizi hızlandırır.")
32
1. sıcak antikor aracılıklı OİHA (IgG bağımlı)
Antikor genellikle Ig G yapısında olup eritrosit memrandaki Ag’lere özellikle Rh antijenlerinden birine karşıdır (spesifik) veya bazen bütün eritrosit panelleri ile reaksiyon verir(panaglutinin). Bu antikorlar sıklıkla 37 derecede optimal aktiviteye sahip olduğu için sıcak (ılık) Ab aracılı otoimmün hemolitik anemi de denir.
 veya bazen bütün eritrosit panelleri ile reaksiyon verir(panaglutinin). Bu antikorlar sıklıkla 37 derecede optimal aktiviteye sahip olduğu için sıcak (ılık) Ab aracılı otoimmün hemolitik anemi de denir.")
33
OİHA – Ig G & Etyoloji Hastaların % 40’ında sekonder bir sebebe bağlıdır. Geri kalanında idiopatiktir. Sekonder nedenler; 1. KLL (***en sık malignite), lenfoma ve diğer Lenfoproliferatif hastalıklar, over teratomu, timoma, karsinomlar (mide) 2. SLE (*en sık kollejenoz hastalık), PSS, RA, ülseratif kolit, kronik aktif hepatit, PAN 3. İlaç (*metil dopa en sık) 4. Enfeksiyonlar (özellikle respiratuvar viral, EMN, CMV, Tbc)
, lenfoma ve diğer Lenfoproliferatif hastalıklar, over teratomu, timoma, karsinomlar (mide) 2. SLE (*en sık kollejenoz hastalık), PSS, RA, ülseratif kolit, kronik aktif hepatit, PAN. 3. İlaç (*metil dopa en sık) 4. Enfeksiyonlar (özellikle respiratuvar viral, EMN, CMV, Tbc)")
34
OİHA – Ig G & Klinik Bütün yaş gruplarında görülebilmekle birlikte en sık erişkin bayanlarda görülür Semptomlar genellikle yavaş başlamakla birlikte (Lenfoma, SLE), ani başlangıçlı olgularda vardır (akut viral infeksiyon). Spenomegalinin(%80) yanısıra HM(%45) ve LAP’da(%35) saptanabilir. Sarılık, solukluk, abdominal ağrı ve ateş görülür.
, ani başlangıçlı olgularda vardır (akut viral infeksiyon). Spenomegalinin(%80) yanısıra HM(%45) ve LAP’da(%35) saptanabilir. Sarılık, solukluk, abdominal ağrı ve ateş görülür.")
35
SPLENOMEGALİ VE TROMBOSİTOPENİ EVANS SENDROMU

36
OİHA – Ig G & Laboratuvar
Anemi: hafif veya ağır olabilir. Direkt coombs ile IgG tipi antikorlar saptanır (+), indirek coombs (+) veya (-) olabilir. İnd AGT (+) olması çok fazla miktarda eritrosit otoantikoru yapılmış olduğunu gösterir. OEH ve OEHbK genellikle artmıştır. OEH bazen 115 fl’nin üzerine çıkabilir. PY’da anizositoz, polikromazi, mikrosferositler, rulo formasyonu, hemoliz ciddi ise normoblastlar görülebilir.
, indirek coombs (+) veya (-) olabilir. İnd AGT (+) olması çok fazla miktarda eritrosit otoantikoru yapılmış olduğunu gösterir. OEH ve OEHbK genellikle artmıştır. OEH bazen 115 fl’nin üzerine çıkabilir. PY’da anizositoz, polikromazi, mikrosferositler, rulo formasyonu, hemoliz ciddi ise normoblastlar görülebilir.")
37
OİHA – Ig G & Laboratuvar
Retikolositoz vardır. Nadiren tanı anında %25 olguda retikülositopeni olabilir. Bu durumda ileri tetkik yapmak gerekir; bu tetkikler arasında infiltratif bir olayı saptayaabilmek için Kİ asp ve biopsisi ve infeksiyöz nedenlere özellikle parvo virüs B19’ ayönelik serolojik testler sayılabilir. İndirekt biluribün ve LDH yüksek, haptoglobulin düşüktür. Yıkım dalakta olduğu için hemosidererinüri yoktur.

38
OİHA – Ig G & Ayırıcı Tanı
Periferik yaymada sferosit görülmesine yol açan durumlarla yapılmalıdır. 1. Herediter sferositoz 2. Zieve sendromu 3. Clostridial sepsis

39
OİHA – Ig G & Tedavi 1. Transfüzyon (cok gerekmedikce yapılmamalı)
2. Steroid ilk tercih edilecek ajandır... İlk 3 hafta içinde steroide anlamlı yanıt alınamayan (%15-20) veya hastalığı kontrol altına almak için gerekli olan prednizon dozu yüksek ise (15 mg/kg/gün üzerinde ) veya nüks varlığında diğer tedavi seçenekleri düşünülmelidir. 3. ikinci aşamada seçilmesi gereken tedavi yöntemi Splenektomidir.. 4. üçüncü aşamada seçilecek tedavi immun supressif ilaçlardır. Azotiopürin siklofosfamid. Bu ilaçlara genellikle cevap çok değişken olup, çok iyi değildir(%30-60). Siklosporin 10 mg/kg/gün ile başlanıp kan düzeyleri takip edilerek, kc ve böbrek fonksiyonları izlenerek ilacın dozu ayalanır. 5. Danazol: 600 mg/gün po, denenebilir. IV gamaglobilin diğer tedavilere ek olarak. Genellikle geçici bir yanıt söz konusudur.
 veya hastalığı kontrol altına almak için gerekli olan prednizon dozu yüksek ise (15 mg/kg/gün üzerinde ) veya nüks varlığında diğer tedavi seçenekleri düşünülmelidir. 3. ikinci aşamada seçilmesi gereken tedavi yöntemi Splenektomidir.. 4. üçüncü aşamada seçilecek tedavi immun supressif ilaçlardır. Azotiopürin siklofosfamid. Bu ilaçlara genellikle cevap çok değişken olup, çok iyi değildir(%30-60). Siklosporin 10 mg/kg/gün ile başlanıp kan düzeyleri takip edilerek, kc ve böbrek fonksiyonları izlenerek ilacın dozu ayalanır. 5. Danazol: 600 mg/gün po, denenebilir. IV gamaglobilin diğer tedavilere ek olarak. Genellikle geçici bir yanıt söz konusudur.")
40
2. Soğuk aglütininlere bağlı OİHA (IgM ilişkili)
Genellikle eritrosit membranındaki i/I antijenlerine karşı üretilen ve düşük ısılarda (en iyi 32 derece) aktif olan IgM tipi antikorlar vardır. Soğuk ortamda eritrositleri aglutine ettikleri için “soğuk aglutininler” adıyla anılırlar. Eritrositler esas olarak karaciğerde sekestre olur ve hemoliz için kompleman aktivasyonuna ihtiyaç vardır. Bu antikorlar normal insanlarda da düşük titrelerde(<1:32) mevcuttur. Soğuk aglutininler soğuk aglutinin sendromu paroksismal soğuk hemoglobinürisi olmak üzere başlıca 2 hastalığa neden olurlar. Bunlarda soğuk aglutinin sendromu bütün OİHA’lerin yaklaşık olarak 1/3’ünü meydana getirir.
 aktif olan IgM tipi antikorlar vardır. Soğuk ortamda eritrositleri aglutine ettikleri için soğuk aglutininler adıyla anılırlar. Eritrositler esas olarak karaciğerde sekestre olur ve hemoliz için kompleman aktivasyonuna ihtiyaç vardır. Bu antikorlar normal insanlarda da düşük titrelerde(<1:32) mevcuttur. Soğuk aglutininler. soğuk aglutinin sendromu. paroksismal soğuk hemoglobinürisi olmak üzere başlıca 2 hastalığa neden olurlar. Bunlarda soğuk aglutinin sendromu bütün OİHA’lerin yaklaşık olarak 1/3’ünü meydana getirir.")
41
OİHA Ig M &Sebepler: 1. *En sık idiopatik olarak görülür.
2. *Mikoplazma pnömonia (en sık sekonder sebep), Ebstein Barr, CMV, sıtma ve tripanozoma gibi enfeksiyonlarda titreler artarlar ancak klinik hemoliz bu durumlarda nadirdir. *B-hücreli lenfoproliferatif hastalıklarda örneğin lenfoma ve KLL’de sekonder olarak görülebilir. Bu durumlarda antikor genellikle monoklonaldır ve elektroforezde monoklonal pik yapar.
, Ebstein Barr, CMV, sıtma ve tripanozoma gibi enfeksiyonlarda titreler artarlar ancak klinik hemoliz bu durumlarda nadirdir. *B-hücreli lenfoproliferatif hastalıklarda örneğin lenfoma ve KLL’de sekonder olarak görülebilir. Bu durumlarda antikor genellikle monoklonaldır ve elektroforezde monoklonal pik yapar.")
42
OİHA Ig M & Klinik Bu tür hemoliz komplemanla olan opsonizasyon sonucu ekstravasküler alanda gerçekleşir. Nadiren de doğrudan lizis ile intravasküler alanda da olabilir. Hastalık soğuk ortamlarda daha sıktır, raynaud benzeri tablo veya akrosiyanoz ile seyredebilir. Klinik tablo genellikle ılımlı bir seyir izler. Anemi ve hafif sarılıkla seyredip diğer laboratuvar bulguları hemolizi gösterir. Soğuk aglutininler eritrositlere yüzeyel damarlarda bağlanarak, kan akımını bozarlar ve akrosiyanoza yola açarlar. Mikrosirkülasyonda oluşan dolaşım bozukluğu sonucunda livedo retikülaris izlenebilir. Bu hastalarda genellikle splenomegali izlenmez.

43
OİHA Ig M & Laboratuar Periferik yaymada otoaglutinasyon tipiktir.
Hastadan alınan kanın hızla pıhtılaşması tipiktir. Klasik coombs (-) çıkabilir ancak anti C3 kullanılarak yapılan Coombs (+) tir (kompleman). Soğuk aglutinin titresi 1/10.000’in üzerinde olması tanı için önemlidir. Retikülositoz Serum haptoglobin azalmış LDH ve ind. Bil düzeyleri artmış
 çıkabilir ancak anti C3 kullanılarak yapılan Coombs (+) tir (kompleman). Soğuk aglutinin titresi 1/10.000’in üzerinde olması tanı için önemlidir. Retikülositoz. Serum haptoglobin azalmış. LDH ve ind. Bil düzeyleri artmış.")
44
OİHA Ig M & Tedavi Tedavi esas olarak altta yatan sebebin tedavisidir.
1. Hastanın soğuktan korunması ve kendisini ılık tutması gereklidir. Bu nedenle yatak istirahati bu hastalığın tedavisinde önemlidir. Daha ılık iklime sahip bölgelerde yaşaması önerilir. 2. İmmünsupresif tedavi: klorambusil ve siklofosfamid kullanılabilir. Klorombusil 2-4 mg/gün, po, başlanıp soğuk aglutinin sentezini kontrol altına alacak şekilde dozu 10 mg/güne kadar artırılabilir. Siklofosfamid vesteroid kombine de kullanılabilir. 3. Steroid ve splenektominin faydası yoktur. 4. Plazmaferez bu hastalarda antikorları uzaklaştırması ile faydalı olabilir. 5. Bu hastalara kan vermek çok tehlikeli olabilir. Transfüzyon şartsa 37 dereceye kadar kanı ısıtmak ve yıkanmış eritrosit suspansiyonu ve geniş bir venden vermek gerekir. Transfüzyon yaparken hasta ısıtılmalı ve transfüzyon yavaş yapılmalıdır.

45
G. İLAÇLARA BAĞLI İMMÜN HEMOLİTİK ANEMİ Hapten tipi:
Bazı ilaçlar eritrosit membranına sıkıca bağlanarak antijenik özellik kazanır ve daha sonra ilaca karşı oluşan antikor eritrosit membranında ilacın olduğu bölgelere bağlanır ve eritrosit yıkımını başlatırlar (örn. Penisilin, sefalosporin). Hemoliz genellikle ekstravaskülerdir. Kompleman aktivasyonu genellikle vardır. Ig G-Direkt coombs (+) dir. İndirekt coombs ise tipik olarak (-) dir.
. Hemoliz genellikle ekstravaskülerdir. Kompleman aktivasyonu genellikle vardır. Ig G-Direkt coombs (+) dir. İndirekt coombs ise tipik olarak (-) dir.")
46
G. İLAÇLARA BAĞLI İMMÜN HEMOLİTİK ANEMİ İmmun kompleks tipi (kinidin tipi):
Eritrosit membranına bağlanan bazı ilaçlar, bir eritrosit antijeni ile birlikte yeni bir antijen oluştururlar. Ilaç+Plazma proteinleri kompleks oluşturduktan sonra ilaca özgü kompleman fikse eden antikorların yapımını uyarırlar. Bu antikorlar Ig G veya Ig M yapısında olabilir. Ab’lar yeni yapıya bağlanır. Eritrosit membranında ilaç- antikor immün kompleksleri meydana geldikten sonra C3b eritrosit membranına bağlanarak alternatif yoldan kompleman sistemini aktive eder (C3 eritrosit yüzeyinde birikir). Kompleman sistemi aktive olduktan sonra hızlı bir şekilde intravasküler eritrosit yıkımı meydana gelir.
. Kompleman sistemi aktive olduktan sonra hızlı bir şekilde intravasküler eritrosit yıkımı meydana gelir.")
47
İlaca bağlı en ağır hemoliz tipidir.
G. İLAÇLARA BAĞLI İMMÜN HEMOLİTİK ANEMİ İmmun kompleks tipi (kinidin tipi): Direkt coombs (+) dir. İndirekt AGT (-) dir. İlaca bağlı en ağır hemoliz tipidir. Ağır intravasküler hemoliz ve sonrasında böbrek yetmezliğine ve DIK’e yol açabilir. Temel ilaç Kinin, kinidin olup rifampin, probenesid, diklofenak ve klorpropamid ile de olabilir.
: Direkt coombs (+) dir. İndirekt AGT (-) dir. İlaca bağlı en ağır hemoliz tipidir. Ağır intravasküler hemoliz ve sonrasında böbrek yetmezliğine ve DIK’e yol açabilir. Temel ilaç Kinin, kinidin olup rifampin, probenesid, diklofenak ve klorpropamid ile de olabilir.")
48
Bazı ilaçlar doğrudan otoantikor yapımını uyarır.
G. İLAÇLARA BAĞLI İMMÜN HEMOLİTİK ANEMİ Otoimmun reaksiyon tipi (metil topa tipi): Bazı ilaçlar doğrudan otoantikor yapımını uyarır. Bu tip IgG bağımlı hemolitik anemi ile patofizyolojik olarak aynıdır. Hemoliz ekstravasküler alanda gerçekleşir. Hemolize neden olan antikorlar metil dopa ile reaksiyona girmezler. Bu Ab’lar eritrositlerin Rh antijenleri ile reaksiyona girerler.
: Bazı ilaçlar doğrudan otoantikor yapımını uyarır. Bu tip IgG bağımlı hemolitik anemi ile patofizyolojik olarak aynıdır. Hemoliz ekstravasküler alanda gerçekleşir. Hemolize neden olan antikorlar metil dopa ile reaksiyona girmezler. Bu Ab’lar eritrositlerin Rh antijenleri ile reaksiyona girerler.")
49
Kronik metil dopa tedavisi alan hastaların %15’inde hemoliz olur.
G. İLAÇLARA BAĞLI İMMÜN HEMOLİTİK ANEMİ Otoimmun reaksiyon tipi (metil topa tipi): Kronik metil dopa tedavisi alan hastaların %15’inde hemoliz olur. Bu antikorlar ayrılırsa metil-dopasız ortamda da normal eritrositleri hemolize eder. Hem direkt hem indirekt coombs (+) dir. İlacın hemen kesilmesi tedavideki temel yaklaşımdır. Ağır olgularda eritrosit suspansiyonu gerekebilir. Bu hastaların çoğunda ANA, RF, mide mukozasına karşı Ab lar da saptanabilir
: Kronik metil dopa tedavisi alan hastaların %15’inde hemoliz olur. Bu antikorlar ayrılırsa metil-dopasız ortamda da normal eritrositleri hemolize eder. Hem direkt hem indirekt coombs (+) dir. İlacın hemen kesilmesi tedavideki temel yaklaşımdır. Ağır olgularda eritrosit suspansiyonu gerekebilir. Bu hastaların çoğunda ANA, RF, mide mukozasına karşı Ab lar da saptanabilir.")
50
PAROKSİSMAL NOKTÜRNAL HEMOGLOBİNÜRİ
Edinilmiş bir kök hücre defekti vardır. İntrensek eritrosit kusuruna bağlı tek edinsel hemolitik hastalıktır. Aslında kronik bir intravasküler hemoliz söz konusu olup dönem dönem agreve olmaktadır. Kompleman bağımlı membran hasarına duyarlı hücrelerin artması ile karekterize bir hastalıktır

51
PAROKSİSMAL NOKTÜRNAL HEMOGLOBİNÜRİ
Normal eritrositmembranında bulunan DAF (CD55), HRF (Homologous Restriction Factor:C8bp) ve MIRL(CD59) gibi glikoproteinler, kompleman bağımlı hemolizi önleyici etki gösterirler. PNH’li hastalarda bu glikoproteinler genetik bir defekte bağlı olarak (somatik mutasyon) azalmıştır ya da yoktur. Bu nedenle eritrositler kompleman aracılığı ile olan hemolize aşırı derecede duyarlı olurlar.
, HRF (Homologous Restriction Factor:C8bp) ve MIRL(CD59) gibi glikoproteinler, kompleman bağımlı hemolizi önleyici etki gösterirler. PNH’li hastalarda bu glikoproteinler genetik bir defekte bağlı olarak (somatik mutasyon) azalmıştır ya da yoktur. Bu nedenle eritrositler kompleman aracılığı ile olan hemolize aşırı derecede duyarlı olurlar.")
52
PNH & Laboratuvar Serum LDH yüksek Serum haptoglobin düzeyleri azalmış
Hemosiderinüri (+) Hbüri genellikle geceleri olur ve sabah idrar rengi koyulaşır. Renal fonksiyon bozuklukları: albüri, hematüri, hipostenüri, tübüler fonksiyon bozukluğu ve kreatinin klirensi azalma, ABY, KBY, ABY genellikle hemolitik ataktan sonra izlenir ve reverzibldir. Kronik hemoglobinüri sebebi ile demir eksikliği anemisi olabilir.
 Hbüri genellikle geceleri olur ve sabah idrar rengi koyulaşır. Renal fonksiyon bozuklukları: albüri, hematüri, hipostenüri, tübüler fonksiyon bozukluğu ve kreatinin klirensi azalma, ABY, KBY, ABY genellikle hemolitik ataktan sonra izlenir ve reverzibldir. Kronik hemoglobinüri sebebi ile demir eksikliği anemisi olabilir.")
53
PNH & Tanı Sukroz hemoliz testi
Duyarlı ama spesifitesi az, taze normal serum sukrozla dilüe edilerek hastanın eritrositleri ile inkübe edildiğinde hemoliz olması, kompleman aktivasyonu hem klasik hem de alternatif yoldan olur. HAM testi En çok kullanılan ve en sipesifik testtir ama duyarlılığı azdır. Hasta eritrositleri asidifiye normal serumla inkübe edildiklerinde hemoliz olmasıdır. İzlenen hemoliz alternatif yolun aktivasyonu ile meydana gelir

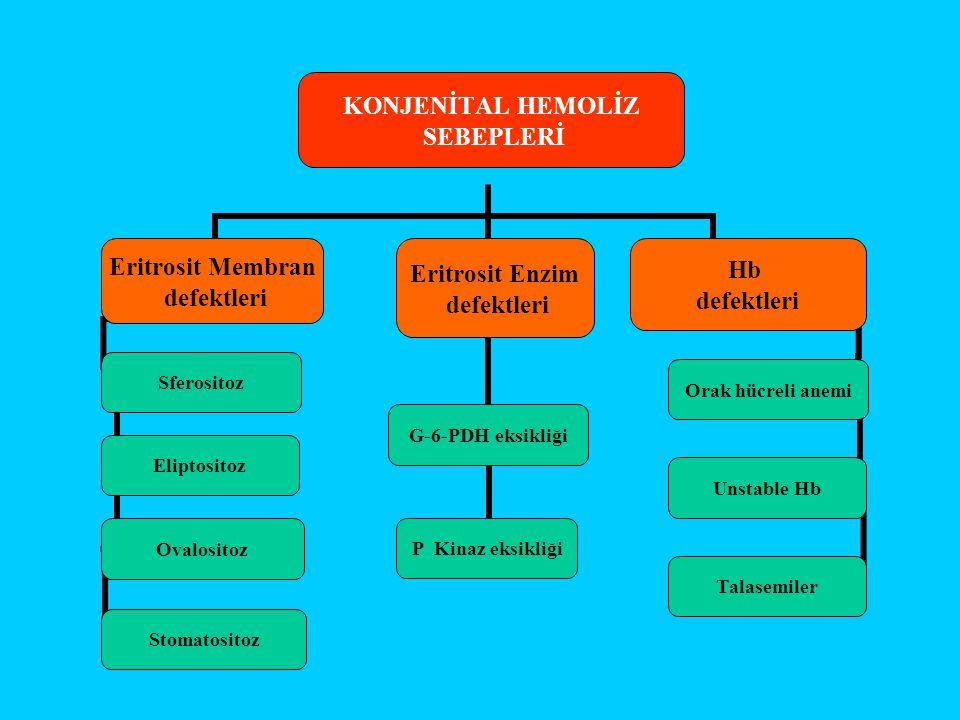
55
A – Eritrosit membran defekti:
Herediter sferositoz, Herediter eliptositoz, Herediter ovalositoz, Herediter stomatositoz Herediter piropoikilositoz

56
HEREDİTER SFEROSİTOZ Otozomal dominant veya resesif geçer
Membran proteinlerinden spektrin veya ankrin de (ankrin defekti daha sık ) bir bozukluk sonucunda….. eritrosit osmotik frajilitesinde artma ve defektif eritrositlerin dalakta hemolizi ile karakterize genetik bir hastalıktır.
 bir bozukluk sonucunda….. eritrosit osmotik frajilitesinde artma ve. defektif eritrositlerin dalakta hemolizi ile karakterize. genetik bir hastalıktır.")
57
Herediter sferositoz

58
HS & Hemolitik kriz: En sık görülen krizdir.
Hayatı tehdit edici değildirler. Genellikle enfeksiyonlara sekonder olur. Hastaların retikülosit sayısında ve indirek bilirubin’de artma görülür. Genellikle günde kendiliğinden düzelir.

59
HS & Aplastik kriz: Nadirdir fakat ağır anemiye neden olduğu için tehlikelidir. Hastalar aplastik kriz sırasında kaybedilebilir. Genellikle parvovirüs B 19 infeksiyonlarına sekonder olarak gelişir. Kİ’de eritroid hipoplazi vardır. Aplastik krizde hastanın Hb/Htk düzeyinde ani düşme ve retikülosit sayısıda azalma izlenir. Hastanın sarılığı kaybolur. Bu dönem yaklaşık 2 hafta sürer. Bu dönemde yapılan transfüzyon hayat kurtarıcı olabilir.

60
HS & Megaloblastik krizler:
Yetersiz folat alımına bağlı yada gebelikte artmış ihtiyaç yüzünden olabilir. Bu nedenle HS’lu hastalara proflaktik folik asit verilmelidir. Bu hastalarda bilurubin safra taşları sıktır. Splenomegali ve sarılık vardır. Gut ve ayak ülserleri olabilir. Hemokromatosis gelişebilir.

61
HS & Tanı-Tedavi Osmotik frajilite testleri kesin tanıyı sağlar.
SDS-PAGE (membran elektroforezi) ile olguların %80’inde memran protein eksiklikleri gösterilebilir. Temel tedavi yaklaşımı Splenektomidir, hastaların çoğunda şifa sağlar. Splenektomi membran defektini değil, anemiyi düzeltir ve krizleri azaltır. Folik asit desteği yapılır.
 ile olguların %80’inde memran protein eksiklikleri gösterilebilir. Temel tedavi yaklaşımı Splenektomidir, hastaların çoğunda şifa sağlar. Splenektomi membran defektini değil, anemiyi düzeltir ve krizleri azaltır. Folik asit desteği yapılır.")
62
PÜRİVAT KİNAZ EKSİKLİĞİ
Kırmızı kürelerde majör ATP sentez yolu EMP’dir. Bu yola ait bir enzim olan pruvat kinaz eksikliğinde dalakta hemoliz olur. Eritrosit 2 – 3 DPG miktarının artması aneminin semptomlarını hafifletebilir. Eksikliğin derecesine göre klinik değişikendir. Splenektomiye kısmen cevap verir.

63
G6PD EKSİKLİĞİ Hekzos monofosfat (pentoz fosfat) yoluna ait bir enzimdir. X’e bağlı genetik geçiş sözkonusudur. En sık enzim eksikliğidir. G-6PD eksik ise eritrositler oksidan strese dayanıksız hale gelir (Redükte glutatyon yokluğunda Hb okside olur. Okside Hb denatüre olur ve Heinz body denilen presipatlat oluşur).
.")
64
G-6PD eksikliği & hemolizi tetikleyen faktörler:
Primakin, Kinakrin, Sulfonamidler (TMP-SMX) Nitrofrantoin, Kloramfenikol, Asetil salisilik asit, Fenasetin (asetominofen bu hastalarda ateş düşürücü olarak kullanılabilir), sulfonlar (Dapson), naftalin, askorbik asit, vit-K, dimerkaprol, probenesid, metilen mayisi, toluidine mavisi Bakla, soya fasülyesi Enfeksiyon (en sık sebep) * İlaçlar: yanda … **** Fenasetin (asetominofen bu hastalarda ateş düşürücü olarak kullanılabilir),
 Nitrofrantoin, Kloramfenikol, Asetil salisilik asit, Fenasetin (asetominofen bu hastalarda ateş düşürücü olarak kullanılabilir), sulfonlar. (Dapson), naftalin, askorbik asit, vit-K, dimerkaprol, probenesid, metilen mayisi, toluidine mavisi. Bakla, soya fasülyesi. Enfeksiyon (en sık sebep) * İlaçlar: yanda … **** Fenasetin (asetominofen bu hastalarda ateş düşürücü olarak kullanılabilir),")
65
ORAK HÜCRELİ ANEMİ (HbS)
Otozomal Resesif geçiş gösterir Yapısal gen bozukluğu ile karakterizedir Beta zincirinin 6. sırasındaki glutamik asit yerine valin gelmiştir. Bu patoloji sonucu eritrositlerin mikrodolaşımdaki esnekliği ve solubilitesi bozulmuştur. Özellikle hipoksi varlığında hücreler oraklaşır, vikositer artar, mikrodamar obstrüksiyonu gelişir.

66
Sickle Cell Anemi – PY Orak hücre Eritroblastlar Howell-Jolly Body

67
HbS & Aplastik krizler;
Parvovirüs B19 enf.eksiyonuna sekonderdir Genellikle sadece bir kez olur (hasta daha sonra bağışıklık geliştirdiği için). Bu yüzen yetişkinlerde nadirdir. Htc düşmesi ile birlikte retikülositin de düşmesi tipiktir. Kan transfüzyonu ile tedavi edilir.
. Bu yüzen yetişkinlerde nadirdir. Htc düşmesi ile birlikte retikülositin de düşmesi tipiktir. Kan transfüzyonu ile tedavi edilir.")
68
TALASEMİLER Hb zincirlerinden birinin veya birkaçının defektif sentezidir. Otozomal resesif geçer. Hipokrom mikrositler bir anemi vardır. Talasemiler ve olarak iki gruba ayrılır.

69
NORMAL ERİŞKİN HEMOGLOBİNLERİ
% 98 HbA (22), % 2-3 HbA2 ( 22) %1-2 HbF (22) vardır.
, % 2-3 HbA2 ( 22) %1-2 HbF (22) vardır.")
70
Alfa talasemi: Gen delesyonu sonucu zincir sentezi azalmıştır.
Normalde 4 alfa globin zinciri geni vardır. Sessiz taşıyıcı → 3 gen varlığında ( x - / xx ) Alfa talasemi taşıyıcı → 2 gen varlığında ( x - / x - ya da -- / xx ) HbH hastalığı → 1 gen varlığında ( - - / - x) Hb Barts → Hiç alfa geni yoksa ( -- / -- ) (4) (hidrops fetallis)
 Alfa talasemi taşıyıcı → 2 gen varlığında ( x - / x - ya da -- / xx ) HbH hastalığı → 1 gen varlığında ( - - / - x) Hb Barts → Hiç alfa geni yoksa ( -- / -- ) (4) (hidrops fetallis)")
71
Alfa talassemia trait

72
BETA TALASEMİ Beta zincir sentezi azalmasıdır.
Beta zincir sentezindeki azalmaya göre heterojen bir grup hastalığı kaplar.

73
Beta thalassemia - heterozygous (minor or trait)
")
74
TALASEMİ MAJÖR (cooleys anemisi)
HbA yok, HbA2 artmış(%4-10), HbF çok artmıştır (%90-96). PY’da; hedef hücreleri, göz yaşı hücreleri, mikro–anizositoz–poikilositoz vardır. Serum Fe , SDBK, kemik iliğinde Fe .
, HbF çok artmıştır (%90-96). PY’da; hedef hücreleri, göz yaşı hücreleri, mikro–anizositoz–poikilositoz vardır. Serum Fe , SDBK, kemik iliğinde Fe .")
75
TALASEMİ İNTERMEDİA Bir miktar HbA üretebilen hastalarda daha hafif seyirli talasemi intermedia gelişir. Bunlarda Hb genellikle 6 – 7 mg/dl’nin üzerindedir. Bunlar yetişkin yaşa ulaşabilir. Talasemilerin tedavisi genellikle destek ve demir şelasyonu ile olmaktadır. Kemik iliği nakli, gen tedavisi ve hemoglomin F sentezini aktive etmeye yönelik tedaviler umut veren deneysel yaklaşımlardır.

76
Beta talasemi Eritrositlerde Bazofilik Stipling

Benzer bir sunumlar