Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Böbreğin konjenital anomalileri ve kistik hastalıkları

2
Normal erişkin

3
Yeni doğan

4
Böbreğin konjenital anomalileri
Tüm insanların %10’u önemli bir ÜS anomalisi ile doğar Çocuklardaki kronik böbrek yetmezliği (KBY) %20 oranında renal displazi ve hipoplaziye bağlıdır
 %20 oranında renal displazi ve hipoplaziye bağlıdır.")
5
Böbreğin konjenital anomalileri
Erişkinlerde KBY’ inin %10’u polikistik böbrek hastalığına bağlıdır Konjenital böbrek hastalığı herediter olabilir, fakat çoğu, gebelikteki kazanılmış gelişimsel defekt sonucudur

6
Tek veya iki taraflı böbrek agenezisi
Üreter tomurcuğunun erken dejenerasyonu sonucu, metanefrik dokunun prolifere olamaması nedendir Tek taraflı 1:1000, iki taraflı 1:3000 sıklıktadır İki taraflı ise intrauterin 14.haftada şiddetli oligohidramnioz vardır. Doğumu takiben birkaç gün içinde ölüm olur

7
Tek veya iki taraflı böbrek agenezisi
İki taraflı olanların %85’i diğer birçok konjenital anomali ile birliktedir (kosta defektleri, hipoplastik akciğerler, vajen ve uterus, vas deferens, seminal veziküllerin yokluğu, kalp anomalileri, trakea ve duodenum atrezisi, yarık dudak damak ve beyin anomalileri gibi). Tek taraflı ise, karşı böbrekte kompansatuar hipertrofi, ileri dönemde ilerleyici glomerüler skleroz ve KBY’ i gelişebilir. Bu durumda da %70’ e kadar ek diğer anomaliler görülebilir
. Tek taraflı ise, karşı böbrekte kompansatuar hipertrofi, ileri dönemde ilerleyici glomerüler skleroz ve KBY’ i gelişebilir. Bu durumda da %70’ e kadar ek diğer anomaliler görülebilir.")
8
Bilateral renal agenesis: Potter sendromu
1/4000 doğumda Oligohidramnioz Potter yüzü: İnfraorbital ve nasolabial katlantılar, gaga burun, büyük-posteriora dönmüş kulaklar İnfantlar, pulmoner hipoplaziye bağlı olarak birkaç gün içinde ölür Oligohidramnioz, akciğer gelişimini bozar ve pulmoner hipoplaziye neden olur

9
Potter(oligohidramnios) görünümü
 görünümü")
10
Oligohidramnios nedenleri
Major nedenler Potter sendromu Bilateral renal displazi Distal ürier yol obstruksiyonu Nadir nedenler İnfantil polikistik böbrek hastalığı Glomerulokistik böbrek hastalığı Renal tubuler disgenesis Kronik amniotik sıvı sızması In utero akut renal yetmezlik İdiopatik

11
Kısmi veya tam üreter dublikasyonu
Üreter tomurcuğunun erken bölünmesi ile oluşur. Metanefrik doku her biri kendi renal pelvisi ve üreterine sahip iki parçaya bölünebilir, İki üreterik tomurcuğun gelişimi sonucu, üreterlerden biri normal pozisyonunu korurken diğeri mezonefrik kanalla birlikte mesanenin alt kısımlarına, üretraya, vajene ve epididimal bölgeye açılabilir.

12
Çift üreterler Bir veya iki üreterin parsiyel veya tam dublikasyonu 150 kişinin 1’ inde görülür Obstruksiyon problemlerine neden olur Böbrekler arasında aorta segmenti bulunmaktadır

13
Böbrek hipoplazisi Böbrekler normal büyüklüğe ulaşamazlar
Bilateral ise erken çocuklukda renal yetmezlik ile sonlanır Unilateral de olabilir Gerçek hipoplazi oldukça nadirdir. Olguların çoğu gelişim yetmezliğinden daha çok, vasküler, enfeksiyöz ve diğer parankimal hastalıklara bağlı olarak gelişir Gerçek hipoplazide, “skar” dokusu olmaz, renal loblar ve piramidler 6 veya daha az sayıdadır ve böbrek arteri de hipoplastikdir Normal bir böbreğin %50’sinden daha küçük olup, displastik elaman içermezler İki tipdir: 1.Basit hipoplazi 2.Oligomeganefroni

14
1.Basit hipoplazi 2.Oligomeganefroni: Bilateral ve non-herediter
1-5 lob vardır ve glomerüller sayıca azalmıştır 2.Oligomeganefroni: Renal hipoplazinin en sık formudur Böbrek küçük, fakat nefronlar çok büyüktür (normalin katı) Konsantrasyon defekti nedeni ile, klinikte jüvenil nefronofitizis ile karışır Proteinüri, böbrek yetmezliği, ilerleyici glomerüler ve tubulointerstisyel fibrozis zaman içerisinde gelişir
 Konsantrasyon defekti nedeni ile, klinikte jüvenil nefronofitizis ile karışır. Proteinüri, böbrek yetmezliği, ilerleyici glomerüler ve tubulointerstisyel fibrozis zaman içerisinde gelişir.")
16
Ektopik böbrek, Böbrek “yükselişi” sırasında umblikal arterlerin oluşturduğu çatal içinden geçemezse, pelvis içinde a.iliaca communis yakınında kalır (En sık pelvik böbrek) İliak, abdominal, subdiafragmatik, supradiafragmatik de olabilir Üreterlerde kıvrımlanma nedeniyle enfeksiyonlara predispozisyon olur
 İliak, abdominal, subdiafragmatik, supradiafragmatik de olabilir. Üreterlerde kıvrımlanma nedeniyle enfeksiyonlara predispozisyon olur.")
17
At nalı böbrek Bazen de arterial çataldan geçerken böbrekler birbirlerine doğru itilir ve kaynaşırlar(At nalı böbrek). Birleşme %90 alt kutupta, %10 üst kutupta olur (Horseshoe Kidney)(1/ ) E lerde sık Turner sendromu ve trisomi 18’ de sık
. Birleşme %90 alt kutupta, %10 üst kutupta olur (Horseshoe Kidney)(1/ ) E lerde sık. Turner sendromu ve trisomi 18’ de sık.")
18
Atnalı "horseshoe" böbrek
Atnalı "horseshoe" böbrek. Diğer anomali veya sendromlar ile birlikte, trisomi 18 gibi, olabilir. İsole anomali şeklinde de de olabilir. Üreterlerin anormal seyrine bağlı olarak parsiyel obstruksiyon sonucu hidronefroz gelişebilir.

19
Böbreğin kistik hastalıkları
Herediter Nonherediter-gelişimsel Kazanılmış 1.klinik, radyolojik, patolojik tanı problemlerine neden olurlar 2.bazı formları, adult polikistik böbrek hastalığı gibi, KBY’inin major sebepleri arasındadır 3.bazen malign tümörler ile karışabilirler

20
Böbrek Kistlerinin sınıflandırması
1. Kistik renal displazi 2. Polikistik böbrek hastalığı a.Otozomal dominant (adult) polikistik hst b.Otozomal resesif (çocukluk) polikistik hst 3. Medüller kistik hastalık a.Medüller sünger böbrek b.Nefronofitizis-üremik medüller kistik hastalık kompleksi 4. Kazanılmış (dializ-birlikte) kistik hastalık 5. Lokalize (basit) renal kist 6. Herediter malformasyon sendromlarında renal kistler (tüberosklerozis gibi) 7. Glomerulokistik hastalık 8.Ekstraparenkimal renal kistler (pyelokaliseal kistler, hilar lenfanjitik kist)
 polikistik hst. b.Otozomal resesif (çocukluk) polikistik hst. 3. Medüller kistik hastalık. a.Medüller sünger böbrek. b.Nefronofitizis-üremik medüller kistik hastalık kompleksi. 4. Kazanılmış (dializ-birlikte) kistik hastalık. 5. Lokalize (basit) renal kist. 6. Herediter malformasyon sendromlarında renal kistler (tüberosklerozis gibi) 7. Glomerulokistik hastalık. 8.Ekstraparenkimal renal kistler (pyelokaliseal kistler, hilar lenfanjitik kist)")
21
Renal disgenesis =Polikistik böbrek hastalığı =renal displazi =multikistik böbrek hastalığı
4 grup Potter Tip I Potter Tip II Potter Tip III Potter Tip IV

22
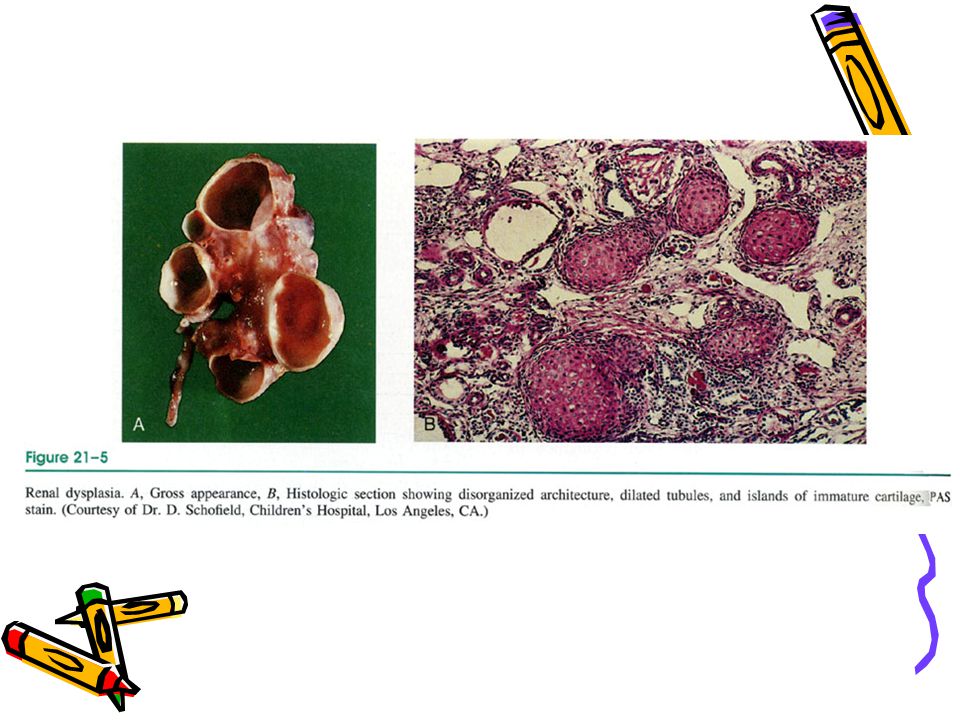
Kistik Renal Displazi Potter Tip II’ ye uyar
Metanefrik diferansiasyonda anomali Genetik kalıtım yoktur Sporadik bir hastalıkdır Mikroskopi: Kıkırdak, andiferansiye mezenşim, immatür toplayıcı duktuslar; anormal lobar organizasyon vardır. Arada normal nefronlar olabilir.

24
Multikistik displastik böbrekler

25
Kistik renal displasi, (Potter tip II),: Geniş kistler, parankimde fibrozis ve kıkırdak dokusu
,: Geniş kistler, parankimde fibrozis ve kıkırdak dokusu")
26
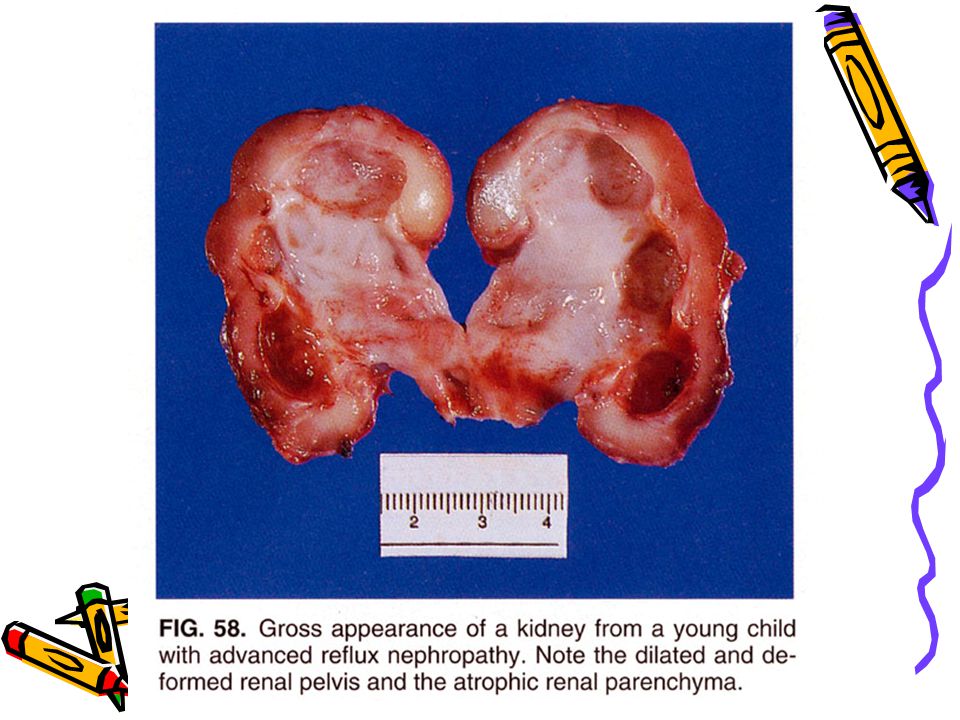
Kistik Renal Displazi Çoğu olguda üreteropelvik obstruksiyon, üreteral agenezis, atrezi, üreterler ve mesane hipoplazisi ve diğer alt üriner yol anomalileri eşlik eder Unilateral veya bilateral olabilir, hemen her zaman kistikdir. Kistler küçük-birkaç cm çapa kadar olabilir, yassı epitel ile döşelidir Tek taraflı ise ağrı nedenidir İki taraflı ise böbrek yetmezliği ile sonlanır

27
Kistik Renal Displazi Multikistik ve aplastik displazi
Segmental displazi Alt üriner yol obstrüksiyonu ile birlikte olan displazi Malformasyon sendromları ile birlikte displazi Herediter renal adisplazi ve ürogenital adisplazi Genellikle otosomal dominant

28
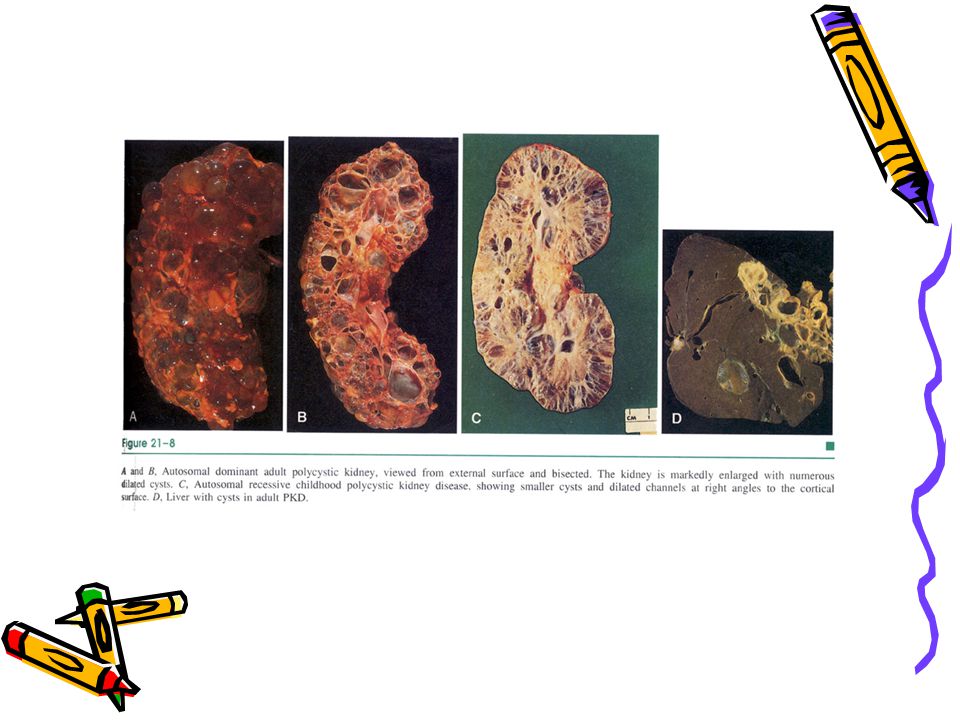
Otozomal dominant (erişkin) polikistik böbrek hst Potter Tip III’ e uyar
Herediter En sık herediter tek gen hastalığıdır 1/ yaşayan doğumda KBY’inin %10’ u Hastalık bilateraldir Renal fonksiyon 4-5. dekada kadar idame edilir. Böbrek yetmezliği ileri yaşlarda belirir

29
Erişkin tip polikistik böbrek hastalığı

30
** Erişkin tip polikistik böbrek hastalığı
Aşağıda ** işaretli olan, normal büyüklükte transplante böbrektir **

31
Erişkin tip polikistik böbrek hastalığı
Ağırlığı 3kg dır

32
Otozomal dominant (ADULT) polikistik böbrek hst
Hastalık sistemikdir. Major patolojik proçes böbreklerdedir. Ekstrarenal konjenital anomaliler: %40’ında polikistik karaciğer hastalığı genellikle asemptomatikdir, kistler bilier epitel ile döşelidir.

33
Karaciğer-mültipl kistler-
Dominant polikistik böbrek hastalığı

34
Otozomal dominant (ADULT) polikistik böbrek hst
Dalak, pankreas, akciğer kistleri, intrakranial Berry anevrizması, mitral valv prolapsusu Ölüm %40 koroner veya hipertansif kalp hst, %25 enfeksiyon, %15 Berry anevrizması rüptürü veya hipertansif intraserebral hemoraji sonucu olur

35
Genetik ve patogenez, 3 ayrı gendeki mutasyonlar ile oluşur
1.PKD1(16p13.3). Gen ürünü “polycystin 1” (bu gendeki mutasyonlar olguların %85’ inden sorumlu) 2.PKD2(4q13-23).Gen ürünü “polycystin 2”(%10) 3.PKD3 Bu proteinler tübüler epitelyal hücre gelişimi ve farklılaşmasında hücre-hücre matriks ilişkilerinde önemlidir Kistleri döşeyen hücrelerde proliferasyon hızı artmıştır, bu hücrelerden sıvı sekresyonu ve anormal ekstraselüler matriks sentezi olur. Sıvıdaki mediatörler ile inflamasyon ve interstisyel fibrozis oluşur. Olaya vasküler hasar da eklenir.
. Gen ürünü polycystin 1 (bu gendeki mutasyonlar olguların %85’ inden sorumlu) 2.PKD2(4q13-23).Gen ürünü polycystin 2 (%10) 3.PKD3. Bu proteinler tübüler epitelyal hücre gelişimi ve farklılaşmasında hücre-hücre matriks ilişkilerinde önemlidir. Kistleri döşeyen hücrelerde proliferasyon hızı artmıştır, bu hücrelerden sıvı sekresyonu ve anormal ekstraselüler matriks sentezi olur. Sıvıdaki mediatörler ile inflamasyon ve interstisyel fibrozis oluşur. Olaya vasküler hasar da eklenir.")
36
Morfoloji Böbrekler bilateral genişlemiştir. Dış yüzden 3-4cm e kadar olabilen, irili ufaklı kistler izlenir. Her bir böbrek 4kg a kadar büyüyebilir Mikroskopide, kistler nefrondaki tübüllerden kaynaklanır ve epitel ile döşelidir. Kist lümeninde berrak bazen hemorajik sıvı bulunur. Kistler arasında fonksiyonel nefronlar görülebilir Klinik: KBY’ i gelişene kadar asemptomatik olabilir. Kanama, kistlerin ilerleyici dilatasyonuna bağlı olarak ağrı, renal kolik ve idrarda hemoraji ile de belirebilir Son dönemde proteinüri, poliüri, hipertansiyon ile karakterli KBY’i gelişir

37
Otozomal resesif (çocukluk) polikistik böbrek hastalığı Potter Tip I’ e uyar
1/ yaşayan doğumda Pulmoner hipoplazi en sık ölüm nedenidir Perinatal, neonatal, infantil ve jüvenil tipleri vardır İlk ikisi çok sıktır, hızla böbrek yetmezliğine gider Böbrekler genişlemiştir, dış yüzleri düzgün görünümdedir Kesit yüzeyinde korteks ve medullada yarık şeklinde kistler vardır. Kistler toplayıcı duktus kökenlidir, distal tubuller de bazen tutulabilir Hastalık bilateraldir Olguların tümünde karaciğerde epitelle döşeli mültipl kistler ve portal safra duktuslarının proliferasyonu vardır Patogenezde kromozom 16 üzerindeki bir gen sorumlu tutulmuştur

38
Ölü fetus, 23, gebelik haftasında oligohidramniosa bağlı akciğer hipoplazi nedeni ile öldü. Hastalık: Oosomal resesif polikistik böbrek hastalığı idi.

39
Otosomal resesif polikistik böbrek hastalığı(ARPKD)
Otosomal resesif polikistik böbrek hastalığı(ARPKD). Glomerül hemen hiç görülmemektedir.
. Glomerül hemen hiç görülmemektedir.")
40
İnfantil ve jüvenil formda karaciğerde periportal fibrozis ve safra duktus proliferasyonu, konjenital hepatik fibrozis, vardır. Daha ileri yaş çocuklarda hepatik tablo baskındır. Splenomegali ile portal hipertansiyon gelişebilir. Bazen, böbrek hastalığı olmadan tek başına veya adult tip polikistik böbrek hastalığı ile birlikte de olabilir

41
Konjenital hepatik fibrozis

43
Renal disgenesis Potter Tip IV
Parsiyel veya aralıklı üretral obstruksiyona ikincil gelişir Retrograd basınç nedeni ile nefron hasarı oluşur Mesane kas hipertrofisi-dilatasyonu, üreterlerin dilatasyonu, hidronefroz ile birliktedir Mesane dilatasyonu Prune-belly sendromu’ nda da olabilir Herediter değildir

44
Prune-belly (Eagle-Barett=triad) sendromu
1/50000 doğumda ve fatal E çocuklarda sık Triad: Karın duvar kasları yok veya hipoplazik İnmemiş testis Üriner yol anomalileri *En erken belirtileri, fetal asit ve Potter sendromu

45
Potter sınıflaması İlgili klinikopatolojik antite
Tip I Tip II Tip III Tip IV İnfantil (otozomal resesif) polikistik böbrek hastalığı, perinatal ve neonatal form Displastik böbrek Erişkin (otozomal dominant) polikistik böbrej Fokal displastik böbrek Herediter sendromlarda displastik böbrek İnfantil polikistik böbrek, infantil ve jüvenil form Alt üriner yol obstruksiyonu ile birliktr displazi
 polikistik böbrek hastalığı, perinatal ve neonatal form. Displastik böbrek. Erişkin (otozomal dominant) polikistik böbrej. Fokal displastik böbrek. Herediter sendromlarda displastik böbrek. İnfantil polikistik böbrek, infantil ve jüvenil form. Alt üriner yol obstruksiyonu ile birliktr displazi.")
46
Renal Displazide multipl malformasyon sendromları
Sık olanlar VATER(VACTERL) birlikte Prune-belly sendromu “Caudal” regresyon sendromu Cloacal ekstrofi Urogenital sinus sendromu Meckel-Grubel sendromu* Dandy-walker sendromu* Kısa kosta-polidaktili sendromu* Elejalde sendromu Nadiren görülenler Trisomi C Trisomi 13 Trisomi 18 Persistan mesonefrik duktus sendromu Zellweger sendromu* Jeune sendromu* Smith-Lemli-Opitz sendromu* Beckwith-Wiedemann sendromu* Laurence-Moon-Bardet-Biedl sendromu* *Otosomal resesif kalıtım gösterenler
 birlikte. Prune-belly sendromu. Caudal regresyon sendromu. Cloacal ekstrofi. Urogenital sinus sendromu. Meckel-Grubel sendromu* Dandy-walker sendromu* Kısa kosta-polidaktili sendromu* Elejalde sendromu. Nadiren görülenler. Trisomi C. Trisomi 13. Trisomi 18. Persistan mesonefrik duktus sendromu. Zellweger sendromu* Jeune sendromu* Smith-Lemli-Opitz sendromu* Beckwith-Wiedemann sendromu* Laurence-Moon-Bardet-Biedl sendromu* *Otosomal resesif kalıtım gösterenler.")
47
İnfantil polikistik böbrek hastalığı
Konjenital hepatik fibrozis (bilier disgenesis) ile birlikte olan kistik böbrek hastalıkları İnfantil polikistik böbrek hastalığı Erişkin polikistik böbrek hastalığı Jüvenil nefronofitizis Meckel-Gruber sendromu* Zellweger sendromu* Ivemark sendromu* Kondrodisplastik sendromlar* Trisomi C* Trisomi D* *Ek malformasyon olanlardır
 ile birlikte olan kistik böbrek hastalıkları. İnfantil polikistik böbrek hastalığı. Erişkin polikistik böbrek hastalığı. Jüvenil nefronofitizis. Meckel-Gruber sendromu* Zellweger sendromu* Ivemark sendromu* Kondrodisplastik sendromlar* Trisomi C* Trisomi D* *Ek malformasyon olanlardır.")
48
Renal medulla’nın kistik hastalıkları
Medüller süngerimsi böbrek Nefronofitizis- Üremik medüller kistik hastalık kompleksi

49
Medüller süngerimsi böbrek
Piramidlerin papiller toplayıcı duktuslarının mültipl kistik dilatasyonları Adultlarda, genelde bilateral ve E’lerde görülür Kistler kübik epitel ile döşelidir Kortikal skarlaşma olmaz Patogenez bilinmemektedir Böbrek fonksiyonu normaldir

50
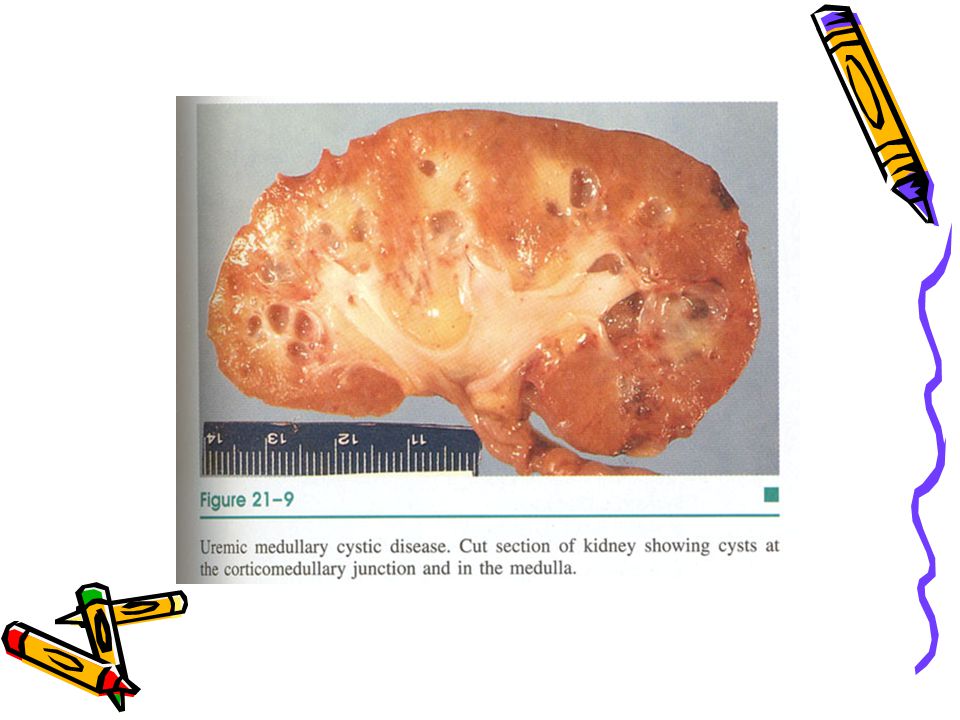
Nefronofitizis-üremik medüller kistik hastalık kompleksi
Genellikle çocuklukta başlayan bir grup ilerleyici böbrek hastalığı, Kortikal tubuler atrofi, interstisyel fibrozis ile birlikte medullada kistler ile karakterlidir Herediter tubulointerstisyel nefrit olarak da bilinir ve böbrek yetmezliği ile sonlanır

51
Nefronofitizis-üremik medüller kistik hst kompleksi (Çocuk ve adolesanlardaki KBY lerinin %20 sinin nedenidir) 4 varyantı tanımlanmıştır: 1.Sporadik, nonfamilial (%20), 2.Familial jüvenil nefronofitizis, otozomal resesifdir, 3.Renal-retinal displazi (%15), otozomal resesif. Retinitis pigmentoza ile birliktedir, 4.Adult başlangıçlı medüller kistik hastalık, otosomal dominant (%15).
, 2.Familial jüvenil nefronofitizis, otozomal resesifdir, 3.Renal-retinal displazi (%15), otozomal resesif. Retinitis pigmentoza ile birliktedir, 4.Adult başlangıçlı medüller kistik hastalık, otosomal dominant (%15).")
52
Nefronofitizis-üremik medüller kistik hst kompleksi
İdrarı konsantre etme yeteneği bozulur İlk olarak poliüri ve polidipsi ile belirir, 5-10 yılda böbrek yetmezliği gelişir 2q13 de mutasyon vardır Morfoloji: Böbrekler küçük, granüler yüzeyli. Distal tubul ve toplayıcı kanal orijinli kortikomedüller kistler iltihabi hücreler ve fibröz doku ile çevrilidir *Açıklanamayan KBY, aile öyküsü ve biopside kronik tubulointerstisyel nefrit olduğunda düşünülmelidir

54
Kazanılmış (dializ-birlikte) kistik hastalık
İnterstisyel fibrozis ve oksalat kristallerinin tübülleri obstrukte etmesi ile oluşur 0.5-2cm çapında, berrak sıvı ile dolu ve yassı bazen hiperplastik epitel ile döşelidir Asemptomatik. Bazen kistler kanar, hematüriye neden olur İnfeksiyon diğer bir komplikasyonudur Uzun süre dializli hst ların %7’sinde kist duvarından böbrek hücreli karsinom gelişebilir

55
Normal büyüklükte, 2cm den daha küçük çaplı kistler içeren böbrekler, kronik dialize bağlı kistik değişiklik. Medüller yağ dokusu da artmıştır.

56
Kronik hemodialize bağlı kistik değişiklik
Kronik hemodialize bağlı kistik değişiklik. Medüller yağ doku belirgin olarak artmıştır.

57
Uzun süreli renal dializde gelişmiş böbrek hücreli karsinom gelişimi ileri derecede nadirdir

58
Basit kist Mültipl veya tek, genellikle kortikal, 1,5,10 cm çapında olabilen kistlerdir Yassı epitel ile döşeli, berrak bir sıvı ile doludur Bazen hemoraji ani distansiyon ve ağrı; hemorajinin kalsifikasyonu ise garip radyografik gölgelere neden olabilir Önemi, böbrek tümörü ile karışabilmesidir. Tümörlerin aksine radyolojik olarak düz konturlu, avasküler, solid değil sıvı ile doludur Semptomsuz ve bası yoksa müdahale edilmez

59
Basit böbrek kistleri 40 yaşından sonra görülürler

60
Basit böbrek kistleri. Multipl olabilir. Yaşlılarda daha sık görülür.

61
Otopside görülen, insidental basit kist

62
Von Hippel-Lindau hastalığı
Otosomal dominant Renal ve ekstrarenal kistler Multipl ve bilateral renal kistler (glikojen zengin hücrelerle döşelidir) Epididim, pankreas kistleri Neoplazmlar Böbrek hücreli karsinom (bilateral ve multipldir) Pankreas kistadenomu Hemanjioblastomlar (Retina, serebellum ve spinal) Feokromositoma
 Epididim, pankreas kistleri. Neoplazmlar. Böbrek hücreli karsinom (bilateral ve multipldir) Pankreas kistadenomu. Hemanjioblastomlar (Retina, serebellum ve spinal) Feokromositoma.")
63
Tuberous sclerosis Otosomal dominant
Mental retardasyon, epilepsi, anjiofibromlar, kardiak rabdomyomlar Böbrekte kistler ve anjiomyolipomlar Böbrek kistleri küçük ve az sayıdadır Çocuklarda otosomal dominant polikistik hastalığı andıran difüz kistler gelişebilir

64
Glomerulokistik hastalık (GKH)
Otosomal dominat GKH Familial hipoplastik GKH Sporadik GKH Temel anomali olabileceği gibi, renal ve ekstrarenal diğer anomalilerle de birlikte olabilir Glomerüler kistlerin olabildiği sendromlar Örn. Tubero sklerosis, trisomi 13

65
Finnish tip konjenital nefrotik sendrom
Otosomal resesif Yaşamın ilk 3 ayındaki nefrotik sendromun en sık nedeni İlerleyici glomerüler skleroz ve tubuler atrofi Toplayıcı duktuslar ve Bowman kapsülü orjinli küçük kistler İnfantlar 3 yıl içinde böbrek yetmezliği ve infeksiyon ile kaybedilirler

66
Renal tubuler disgenesis
Tubul diferansiasyonunun yetersizliği Özellikle distal tubul ve toplayıcı duktuslar Oligohidramnios, Potter sendromu ve hipoplastik akciğer ile karakterli Otosomal resesiftir

Benzer bir sunumlar