Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
M. A. Göz M: Normal 11 yaş, erkek İlk başvuru: 4 yaş
Gözlerde şişlik, ateşlenme, ara ara eklem ağrısı Akrabalık yok. Annede üreterde darlık: opere Enürezis nokturna, sekonder enkoprezis, boy kısalığı(BH tdv almış) Biyokimya: Glu:88, Üre: 45, Cre: 0.55, ÜA:2.7, Na:140, K: 3.9, Cl:102, Ca: 9.3, P:2.8, T.prot: 8.2, Alb: 4.6 Hemogram normal TIT: ph:6.5-7, Dans: , 7-8 eritrosit 0.5-1 g/gün proteinüri 7.7 mg/kg/gün kalsiüri Renal USG: Bilateral nefrolitiazis, G2 parankim hast. DMSA: KBY ile uyumlu Böbrek bx: Bazal memb: N, Mezangial matrix orta derece artmış(elektron yoğun bölgeler), ep ayaksı uzantılarda kısmi ob, tub lümen ve inters. Lenfosit ve granülositler Göz M: Normal
 Biyokimya: Glu:88, Üre: 45, Cre: 0.55, ÜA:2.7, Na:140, K: 3.9, Cl:102, Ca: 9.3, P:2.8, T.prot: 8.2, Alb: 4.6. Hemogram normal. TIT: ph:6.5-7, Dans: , 7-8 eritrosit g/gün proteinüri. 7.7 mg/kg/gün kalsiüri. Renal USG: Bilateral nefrolitiazis, G2 parankim hast. DMSA: KBY ile uyumlu. Böbrek bx: Bazal memb: N, Mezangial matrix orta derece artmış(elektron yoğun bölgeler), ep ayaksı uzantılarda kısmi ob, tub lümen ve inters. Lenfosit ve granülositler. Göz M: Normal.")
2
A. A. Göz M: Normal 16 yaş, erkek İlk başvuru: 4.5 yaş
Bulanık idrar yapma, idrar yolu infeksiyonu 3 yaşında pyelonefrit tanısı ile yatış, DKÇ öyküsü Akrabalık yok. Annede üreterde darlık: opere Sistemik muayene N, büyüme gelişme N Biyokimya: Glu:92, Üre: 30, Cre: 0.87, ÜA:2.8, Na:143, K: 3.9, Cl:100, Ca: 9.5, P:2.8, T.prot: 7.5, Alb: 4.5 Hemogram normal TIT: ph:5-7.5, Dans: , eritrosit g/gün proteinüri Renal USG: Bilateral renal parankim ekojenitesinde min. Artış, kalisiel yapılarda kistik genişleme( Meduller kistik hast?) VCUG: Normal DMSA: KBY ile uyumlu Böbrek bx: Hafif mez. artış, seyrek global skleroz, fokal tubuler atrofi Göz M: Normal
 VCUG: Normal. DMSA: KBY ile uyumlu. Böbrek bx: Hafif mez. artış, seyrek global skleroz, fokal tubuler atrofi. Göz M: Normal.")
3
U. K. 13 yaş, erkek İlk başvuru: 12 yaş
Nefrolitiazis, hipofosfatemik rahitis tanıları ile 12 yıldır takipli Göz operasyonu (10 günlük, ety?) Akrabalık (+). İki erkek kardeş, birinde konj. Katarakt ve glokom var Boy kısalığı, O-bine bacak deformitesi Biyokimya: Glu:95, Üre:36, Cre: 0.93, ÜA:5.6, Na:142, K: 3.5, Cl:104, Ca: 10.3, P:2.8, ALP: 264, T.prot: 8.2, Alb: 4.8 Hemogram normal Kan gazı: pH: 7.35, HCO3: 20, BE: -4.9, pCO2 : 36.3 TIT: ph:5-6, Dans: , 7-8 eritrosit, bol hyalen silendir, bol amorf fosfat 2.7 g/gün proteinüri 6 mg/kg/gün kalsiüri Renal USG: Sağ böbrekte nefrolitiazis, sol böbrekte nefrokalsinozis Göz M: Sistin kristali yok.
 Akrabalık (+). İki erkek kardeş, birinde konj. Katarakt ve glokom var. Boy kısalığı, O-bine bacak deformitesi. Biyokimya: Glu:95, Üre:36, Cre: 0.93, ÜA:5.6, Na:142, K: 3.5, Cl:104, Ca: 10.3, P:2.8, ALP: 264, T.prot: 8.2, Alb: 4.8. Hemogram normal. Kan gazı: pH: 7.35, HCO3: 20, BE: -4.9, pCO2 : TIT: ph:5-6, Dans: , 7-8 eritrosit, bol hyalen silendir, bol amorf fosfat. 2.7 g/gün proteinüri. 6 mg/kg/gün kalsiüri. Renal USG: Sağ böbrekte nefrolitiazis, sol böbrekte nefrokalsinozis. Göz M: Sistin kristali yok.")
4
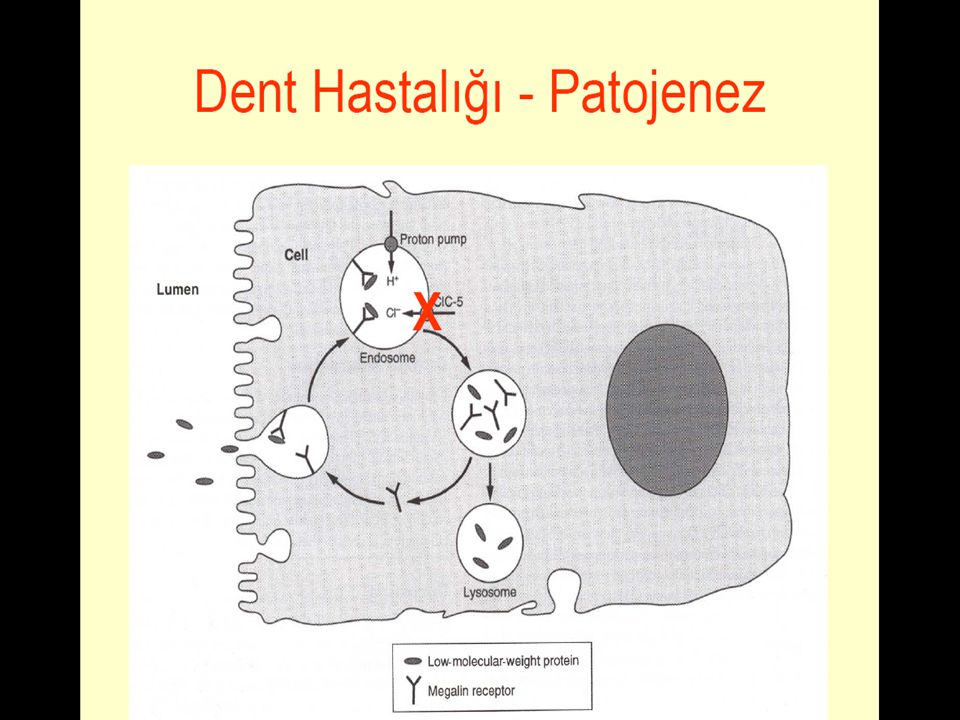
DENT HASTALIĞI Düşük moleküler ağırlıklı proteinüri ve nefrokalsinoz
X-bağlı resesif hipofosfatemik rahitis +/-hiperkalsiüri X-bağlı resesif nefrolitiaz Gen sembol: CLCN5 Kromozomal bölge: Xp11.22 Protein adı: Chloride channel protein 5 Gen sembol: OCRL Kromozomal bölge: Xq26.1 Protein adı: Inositol polyphosphate 5-phosphatase OCRL-1 Dent Disease with mutations in OCRL1. Am J Hum Genet (2005) 76: 260-7
 76:")
5
DENT HASTALIĞI İlk kez 1964’ te Dent and Friedman, hiperkalsiüri, hiperfosfatüri proteinüri ve aminoasidüri ile birlikte olan 2 akraba olmayan ingiliz erkek rikets olgusu bildirdiler Frymoyer ve arkadaşları 1991’ de X’ e bağlı geçen nefrolitiazise ait gen buldular (NPHL1 and NPHL2) Lloyd ve ark. 1996’ da 8 ingiliz ailede CLCN5 geninde mutasyon saptadılar Hoopes ve ark. 2005’ te 13 vakalık bir seride OCRL1 gen mutasyonunu gösterdiler.
 Lloyd ve ark. 1996’ da 8 ingiliz ailede CLCN5 geninde mutasyon saptadılar. Hoopes ve ark. 2005’ te 13 vakalık bir seride OCRL1 gen mutasyonunu gösterdiler.")
6
Klinik Bulgular Boy kısalığı Genitoüriner:
-Proksimal renal tubuler defekt -Azalmış fosfor reabsorbsiyonu -Nefrokalsinozis -Nefrolitiazis -Progresif renal yetersizlik -Erişkin dönemde renal yetmezlik İskelet Sistemi -Rahitis (%33) -Osteomalazi -Kemik ağrıları
 -Osteomalazi. -Kemik ağrıları.")
7
Laboratuvar Düşük moleküler ağırlıklı proteinüri Hiperkalsiüri
Hipofosfatemi Hiperfosfatüri Aminoasitüri Glikozüri Mikroskopik hematüri 1.25 dihidroksi D3 yüksekliği

8
Dent Hastalığı ve Lowe Sendromunun Renal Bulguları
Dent hastalığı için 2 gen bildiriliyor: CLCN5, OCRL1 Dent H. ve Lowe S. Tanısı konmuş hastalarda fenotip-genotip korelasyon var mı? 12 akraba olmayan fenotip olarak Dent H. İle uyumlu erkek hasta -CLCN5 mutasyonu hasta (Dent hastalığı 1) -OCRL1 mutasyonu hasta (Dent hastalığı 2) -Bir hastada bu iki gen mutasyonu saptanamamış. Lowe S. fenotipindeki 7 hastanın yedisinde de OCRL1 mut (+) Lowe S. Hastalarda hipofosfatemik rikets ve tubuler proteinüri Dent 1 hastalarından daha fazla bulunmuş Dent 2 Hastalarında, Dent 1’ e göre daha yüksek dereceli proteinüri ve hiperkalsiüri saptanmış.
 -OCRL1 mutasyonu 2 hasta (Dent hastalığı 2) -Bir hastada bu iki gen mutasyonu saptanamamış. Lowe S. fenotipindeki 7 hastanın yedisinde de OCRL1 mut (+) Lowe S. Hastalarda hipofosfatemik rikets ve tubuler proteinüri Dent 1 hastalarından daha fazla bulunmuş. Dent 2 Hastalarında, Dent 1’ e göre daha yüksek dereceli proteinüri ve hiperkalsiüri saptanmış.")
9
Dent Hastalığı XLR LMWP, hiperkalsiüri, glikozüri, aminoasitüri, fosfatüri olan proksimal tubulopatidir. Nefrokalsinozis, nefrolitiazis, progresif renal yetm, hipofosfatemik rikets gelişebilir. Bu hastalarda tipik bir pRTA bulguları ya da ekstrarenal bulgular olmayabilir. CLCN5 gen ; Xp11.22, renalklorid/proton porter(CIC-5) ı kodlar 2005 tarihinde OCRL1 gen mutasyonu bildirilerek genetik heterojenite vurgulandı. OCRL1 geni; trans-Golgi ağındaki PIP2’ ı kodlar.
 ı kodlar tarihinde OCRL1 gen mutasyonu bildirilerek genetik heterojenite vurgulandı. OCRL1 geni; trans-Golgi ağındaki PIP2’ ı kodlar.")
11
Lowe Sendromu XLR Bilateral konjenital katarakt Renal Fankoni sendromu
Mental retardasyon

12
Dent hastalığı için klinik kriterler:
1) LMW proteinüri 2) Hiperkalsiüri 3) Lowe S diagnostik kriterlerinin olmaması -Bilateral konjenital katarakt -Renal Fankoni send. -Mental retasdasyon
 LMW proteinüri. 2) Hiperkalsiüri. 3) Lowe S diagnostik kriterlerinin olmaması. -Bilateral konjenital katarakt. -Renal Fankoni send. -Mental retasdasyon.")
13
Table 1 Definitions of renal manifestations
Description LMW proteinuria At least fivefold above the upper limit of the normal level of the urinary β2 microglobulin:creatinine ratio Hypercalciuria Urinary calcium excretion higher than age-matched normal values [13] and/or nephrocalcinosis/nephrolithiasis on renal ultrasonographic examinations Microscopic hematuria Presence of five or more red blood cells per high power field observed during microscopic examination of urine Hypophosphatemia A serum phosphate concentration lower than the age-matched normal level and tubular reabsorption of phosphate < 85% Hyperuricosuria A serum uric acid concentration < 3.0 mg/dL and a fractional excretion of uric acid >10% Glycosuria Two or more episodes of ≥ 1(+) glycosuria as determined by urine dipstick examinations, without hyperglycemia Renal tubular acidosis Persistent hyperchloremic metabolic acidosis with a normal anionic gap, which requires alkali supplement therapy Chronic renal insufficiency A serum creatinine ≥ 1.2 mg/dL for more than 3 months Rickets Typical rachitic changes in bone X-ray studies
 glycosuria as determined by urine dipstick examinations, without hyperglycemia. Renal tubular acidosis. Persistent hyperchloremic metabolic acidosis with a normal anionic gap, which requires alkali supplement therapy. Chronic renal insufficiency. A serum creatinine ≥ 1.2 mg/dL for more than 3 months. Rickets. Typical rachitic changes in bone X-ray studies.")
14
SONUÇLAR Ortalama hastalık başlangıç yaşı: 6.4 yaş (1-13 yaş)
Presantasyon şekilleri -Üriner anomali (okul taramalarında, 6 olgu) -Tesadüfi saptanan proteinüri (5 olgu) -Raşitizm nedeni ile genu varum (1 olgu) Genetik analizden önce 5 olguda böb. Bx: normal CLCN5 gen mutasyonu taşıyan 9 çocukta 7 farklı mutasyon(+) CLCN5 gen mutasyonu taşımayan 3 çocuğun 2 sinde iki farklı OCRL1 mutasyonu, 1 inde mutasyon saptanmadı. CLCN5 veya OCRL1 genlerinde herhangi bir mutasyon olmayan hastada 3,5 yaşında hipofosfatemik raşitizm görüldü,serum bikarbonat değerleri alkali replasmanı yapılmadan mmol/L aralığında dalgalandı, sınırda asidoz görüldü, proksimal tubulopati gelişti. Buna rağmen bu vakada Lowe Sendromu’nun diğer bulgularından olan katarakt ve mental gerilik görülmedi.
 -Tesadüfi saptanan proteinüri (5 olgu) -Raşitizm nedeni ile genu varum (1 olgu) Genetik analizden önce 5 olguda böb. Bx: normal. CLCN5 gen mutasyonu taşıyan 9 çocukta 7 farklı mutasyon(+) CLCN5 gen mutasyonu taşımayan 3 çocuğun 2 sinde iki farklı OCRL1 mutasyonu, 1 inde mutasyon saptanmadı. CLCN5 veya OCRL1 genlerinde herhangi bir mutasyon olmayan hastada 3,5 yaşında hipofosfatemik raşitizm görüldü,serum bikarbonat değerleri alkali replasmanı yapılmadan mmol/L aralığında dalgalandı, sınırda asidoz görüldü, proksimal tubulopati gelişti. Buna rağmen bu vakada Lowe Sendromu’nun diğer bulgularından olan katarakt ve mental gerilik görülmedi.")
15
Table 2 Clinical and genetic features of 12 patients with a clinical diagnosis of Dent disease (D1–D12) and 7 patients with a clinical diagnosis of Lowe syndrome (L1–L7) Patient number Onset (years) Family history Manifestations Mutation in Renal Neurological Others CLCN5 OCRL1 D1 7.8 XR 1,2,3,4 (−) p.C101Ya n.d. D2 8.8 De novo 1,2,4,6 p.S545Na D3 13 1,2,4,7 p.W95X D4 0.2 1,2,3,4,6 p.E609X D5 9 1,2,3 p.R637X D6 6.5 D7 2.8 D8 6.3 c.444insCa D9 3.5 1,2,3,6 c.2141insAa D10 0.1 (+)b c.455delAa D11 6.9 p.F226Sa D12 1,2,3,4,5,6,7 L1 1,2 p.S374Pa L2 0.3 1,3 p.Q260Ea L3 1,5,6,7 Cryptorchidism, thrombocytopenia p.R646X L4 0.7 1,3,4,5,6 Inguinal hernia p.R678X L5 6 n.a. 1,2,4,5,6 L6 1,2,5,7,8 p.R805X L7 1.4 1,2,3,5 Dysplastic testes c.2117del8a
 Family history. Manifestations. Mutation in. Renal. Neurological. Others. CLCN5. OCRL1. D XR. 1,2,3,4. (−) p.C101Ya. n.d. D De novo. 1,2,4,6. p.S545Na. D ,2,4,7. p.W95X. D ,2,3,4,6. p.E609X. D ,2,3. p.R637X. D D D c.444insCa. D ,2,3,6. c.2141insAa. D (+)b. c.455delAa. D p.F226Sa. D12. 1,2,3,4,5,6,7. L1. 1,2. p.S374Pa. L ,3. p.Q260Ea. L3. 1,5,6,7. Cryptorchidism, thrombocytopenia. p.R646X. L ,3,4,5,6. Inguinal hernia. p.R678X. L5. 6. n.a. 1,2,4,5,6. L6. 1,2,5,7,8. p.R805X. L ,2,3,5. Dysplastic testes. c.2117del8a.")
16
Lowe Sendromu klinik tanısı alan 7 hastanın yaş ortalaması 8 ay idi(2ay- 6 yaş).
Bunlardan 3’ü, kriptorşidi, displastik testis, inguinal herni gibi ürolojik bulgular gösterdiler, 1 tanesinde kronik trombositopeni görüldü. 7 hastada, OCRL1 geninde açıklanan 6 farklı mutasyon analiz edildi

17
Table 3 Comparison of renal phenotypes between Dent disease and Lowe syndrome
Renal manifestation Dent disease with mutations in Lowe syndrome (n = 7) P value* CLCN5 (n = 9) OCRL1 (n = 2) None (n = 1) Onset age (years) 6.5 (0.2∼13) 0.1, 6.9 3 0.7 (0.2∼6) 0.012 Urine β2-MG/Cr (ug/mg) 16.2 ± 9.4 43.6, 22.4 12.6 44.7 ± 14.4 0.003 Urine Ca/Cr (mg/mg) 0.43 ± 0.15 1.22, 1.02 0.87 1.16 ± 1.21 > 0.05 Nephrocalcinosis/lithiasis 5/9 (56%) 1/2 (50%) (+) 3/7 (43%) Renal tubular acidosis 0/9 0/2 (−)a 7/7 (100%) 0.000 Hypophosphatemia 6/7(86%) 0.002 Rickets Hyperuricosuria 2/2 6/7 (86%) Glycosuria 1/9 (11%) 2/7 (29%) Hematuria 6/9 (67%) 4/7 (57%) Chronic renal insufficiency (−) 1/7 (11%) Elevated serum CK/LDH n.d. 1/1 3/4 (75%) MG microglobulin, Cr creatinine, Ca calcium, CK creatine kinase, LDH lactate dehydrogenase * Comparison between patients with Dent disease associated with CLCN5 mutations and patients with Lowe syndrome using the Mann–Whitney U test aThis patient had borderline acidosis as indicated by a serum bicarbonate level of 18∼24 mmol/L without alkali supplementation.
 P value* CLCN5 (n = 9) OCRL1 (n = 2) None (n = 1) Onset age (years) 6.5 (0.2∼13) 0.1, (0.2∼6) Urine β2-MG/Cr (ug/mg) 16.2 ± , ± Urine Ca/Cr (mg/mg) 0.43 ± , ± > Nephrocalcinosis/lithiasis. 5/9 (56%) 1/2 (50%) (+) 3/7 (43%) Renal tubular acidosis. 0/9. 0/2. (−)a. 7/7 (100%) Hypophosphatemia. 6/7(86%) Rickets. Hyperuricosuria. 2/2. 6/7 (86%) Glycosuria. 1/9 (11%) 2/7 (29%) Hematuria. 6/9 (67%) 4/7 (57%) Chronic renal insufficiency. (−) 1/7 (11%) Elevated serum CK/LDH. n.d. 1/1. 3/4 (75%) MG microglobulin, Cr creatinine, Ca calcium, CK creatine kinase, LDH lactate dehydrogenase. * Comparison between patients with Dent disease associated with CLCN5 mutations and patients with Lowe syndrome using the Mann–Whitney U test. aThis patient had borderline acidosis as indicated by a serum bicarbonate level of 18∼24 mmol/L without alkali supplementation.")
18
Dent Hastalığı ve Lowe Sendromu tanılı hastaların böbrek tutulumları karşılaştırıldı.
İzlem boyunca Dent hastalarından hiçbirinde renal fonksiyon yetersizliği görülmedi. Ailesinde erkek erişkin Dent hastası olan 4 vakanın, ailedeki hasta bireylerin 2’si 40 yaşında, biri 30 yaşında, diğeri 60 yaşında idi. Masif tubuler proteinüri ve hiperkalsiüriye rağmen etkilenmiş bu aile üyelerinin böbrek fonksiyonları normal idi

19
Lowe Sendromu olan 1 hasta ( Tablo 2’de L6 no’lu) 8 aylıkken böbrek yetersizliği geliştirdi( serum kreatinin 1,4 mg/dl) Lowe Sendromlu hastaların hiçbirinde ailelerinde Lowe Sendromu özellikleri veya kronik böbrek yetersizliği olan erkek erişkin birey yoktu. Lowe Sendromu hastalarında hipofosfatemi ve raşitizm Dent Hastalığı tip 1’den daha sık ve düşük molekül ağırlıklı proteinüri daha belirgin saptandı. Ek olarak, düşük molekül ağırlıklı proteinüri ve hiperkalsiüri derecesi, Dent Hastalığı tip 2’de Tip1’den yüksek ve Lowe Sendromu ile benzer saptandı.

20
Çalışmada Dent Hastalığı tip 2 olgu sayısı az olduğundan istatik analizler yapılamadı. Aynı nedenle Dent Hastalığı tip 1 ve tip 2 olguları diğer renal bulguların sıklığı açısından karşılaştırılmadı

21
Tartışma Dent hastalığının genetik heterojen oluşunu ve Lowe sendromun fenotipik çeşitliliğini saptadık Dent hastalığı teşhisi konulan 12 hastayı klinik olarak değerlendirdik ve 9’unda CLCN5 gene ait mutasyon olduğunu, 2’sinde OCRL1 gende mutasyon olduğunu, ve 1’de de hiçbir gene ait mutasyon olmadığını bulduk Üç benzer çalışmada CLCN5 mutasyonlu hastalarda hastalık insidensi daha yüksek ve her iki gende de mutasyon olmayan hastalarda ise daha düşük olduğunu bildirilmiştir.

22
Table 4 Reported incidences of CLCN5 and OCRL1 mutations in patients clinically diagnosed with Dent disease Patient nationality Total no. of probands No. of probands with mutations in Reference CLCN5 OCRL1 None American, mainly 32 19 (59%) 5 (16%) 8 (25%) [6, 8] Central European, mainly 35 18 (51%) 5 (14%) 12 (34%) [9] Japanese 18 10 (56%) 3 (17%) 5 (28%) [10] 10 6 (60%) n.d. [7] Italiana 24 (group A) 21 (88%) [15] 16 (group B) 3 (19%) Korean 12 9 (75%) 2 (17%) 1 (8%) This study n.d. not done aIn this study, the diagnostic criteria of Dent disease were: (1) low-molecular-weight proteinuria, (2) hypercalciuria, and (3) at least one of the following: nephrocalcinosis, kidney stones, hypophosphatemia, renal failure, aminoaciduria, rickets, or a positive family history, and the patients were grouped into two groups: A, which included those who met all three criteria, and B, which included those who met only two of the three criteria
 5 (16%) 8 (25%) [6, 8] Central European, mainly (51%) 5 (14%) 12 (34%) [9] Japanese (56%) 3 (17%) 5 (28%) [10] (60%) n.d. [7] Italiana. 24 (group A) 21 (88%) [15] 16 (group B) 3 (19%) Korean (75%) 2 (17%) 1 (8%) This study. n.d. not done. aIn this study, the diagnostic criteria of Dent disease were: (1) low-molecular-weight proteinuria, (2) hypercalciuria, and (3) at least one of the following: nephrocalcinosis, kidney stones, hypophosphatemia, renal failure, aminoaciduria, rickets, or a positive family history, and the patients were grouped into two groups: A, which included those who met all three criteria, and B, which included those who met only two of the three criteria.")
23
Tartışma-1 Çalışmamıza dahil edilen hasta sayısı doğru bir yorum için az Dent Hastalığı olan hastaların dahil edilişinde tercih edilen farklı kriterler buna sebep olmuş olabilir. Italyan çalışma Dent hastalığının üç farklı diagnostik kriterini benimsemiştir; LMW proteinuri, hiperkalsiuri ve nephrokalsinozis, böb. taşı, hipofosfatemi, renal yetersizlik, aminoasiduri, rikets, pozitif aile öyküsü bulgularından biri. Bu çalışmada bu üç kriterin hepsini taşıyan 24 hastanın 21’inde CLCN5 mutations fark edilirken (87.5%) Kriterlerin sadece 2’sini taşıyan 16 hastanın 3’ünde (18.8%) mutasyon saptanmış. Çalışmamızın bulgularına benzer şekilde, daha önce yapılan çalışmalar Dent hastalığının tipik fenotipik özelliklerini taşıyan bazı hastalarda CLCN5 VE OCRL1 gen mutasyonu saptanamamıştır. Bu sonuçlar Dent disease’in gelişiminden sorumlu olan üçüncü bir geni öne sürmektedir
 Kriterlerin sadece 2’sini taşıyan 16 hastanın 3’ünde (18.8%) mutasyon saptanmış. Çalışmamızın bulgularına benzer şekilde, daha önce yapılan çalışmalar Dent hastalığının tipik fenotipik özelliklerini taşıyan bazı hastalarda CLCN5 VE OCRL1 gen mutasyonu saptanamamıştır. Bu sonuçlar Dent disease’in gelişiminden sorumlu olan üçüncü bir geni öne sürmektedir.")
24
Tartışma-2 Dent diasease ve Lowe syndrome’un her ikisi de proximal tubular disfonksiyonun benzer belirtilerini içermesine rağmen, disfonksiyonlarının özellikleri birebir aynı değildir. En dikkat çekici farklılık, Lowe sendromun belli başlı belirtilerinden biri olan, Dent hastalığında renal tubular asidosisin yokluğudur. LMW proteinuri her iki hastalıkta da bulunur.

25
Tartışma-3 Proximal tubular disfonsiyona bağlı renal glikozüri, aminoasidüri ve fosfatüri, Lowe sendromunda Dent hastalığına oranla daha sık görülmektedir. Çalışmamızda, Lowe syndrome olan hastalar Dent hastalığı 1 olan hastaların gösterdiğinden daha sık hipofosfatemik rikets ve daha belirgin tubular proteinuri göstermiştir. Dent disease 2 olan hastalar, Dent disease 1 olan hastalara göre tubular proteinuri ve hiperkalsiuri daha yüksek derecede bulundu Progresif renal yetersizlik her iki hastalıkta da yaygın olsa da, Dent hastalarında KBY geç yetişkinlikte gelişirken Lowe sendromlu hastalarda genellikle 10 ile 30 yaşları arasında gelişmektedir Eski çalışmalar hiperkalsiüri, nephrolithiazis bulgularının Dent Hastalığında olduğunu, urinary calsiyum excretionun Lowe sendromunda normal olduğunu bildirmekte iken, yeni çalışmalar hiperkalsiüri ve nefrokalsinozisli hastaların sıklıkla Lowe Sendromlu hastalar olduğunu göstermiştir.

26
Bu çalışmada, Dent hastalığında genetik heterojenite, Lowe sendromunda fenotipik heterojenite tespit edildi. Hafif kas enzimleri yüksekliği, bilişsel-davranışsal gerilik, sistemik asidoz olmaksızın byüme geriliği, kriptorşidi gibi ürogenital anomaliler gibi ekstrarenal bulgular daha çok Dent hastalığı 2 ye işaret eder. Dent hastalığının gelişmesinden sorumlu olabilecek üçüncü bir genin olup olmadığını anlamak için daha fazla çalışma yapılmalıdır. Dent hastalığı ve Lowe sendromundaki fenotip–genotip bağın hala daha açıklanmaya ihtiyacı vardır.

Benzer bir sunumlar












>")






