Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
PRION Dr. Suna Gedikoğlu

2
TARİHÇE

3
1730lar İlk Scrapie olguları 1982 Bu bir protein!! 1950ler
Kuru salgını 1960lar 1985 Bulaşıcılık! Genlerimizde bulunuyor!! 1980ler 1986 Tıbbi Felaket!! SALGIN!!! Deli dana hastalığı 1996 Deli danalı etin insan için önemi

4
PRION HASTALIKLARI Yavaş virus enfeksiyonları
Transmissible Spongiform Encepholopathies (TSE) İnsan ve hayvanda Merkezi Sinir Sistemi’nin etkilendiği bir hastalık grubudur Sporadik Familial Kazanılmış
 İnsan ve hayvanda Merkezi Sinir Sistemi’nin etkilendiği bir hastalık grubudur. Sporadik. Familial. Kazanılmış.")
5
PRION HASTALIKLARI (Transmissible Spongiform Encepholopathies)
Sporadik Familial Kazanılmış Sporadik Creutzfeldt-Jacob Disease (sCJD) Sporadik Familial Insomnia (sFI) Familial Creutzfeldt-Jacob Disease (fCJD) Fatal Familial Insomnia (fFI) Gerstmann-Straussler-Scheinker Syndrome (GSS) İyatrojenik Creutzfeldt-Jacob Disease (iCJD) Varyant Creutzfeldt-Jacob Disease (vCJD) Kuru
 Sporadik Familial Insomnia (sFI) Familial Creutzfeldt-Jacob Disease (fCJD) Fatal Familial Insomnia (fFI) Gerstmann-Straussler-Scheinker Syndrome (GSS) İyatrojenik Creutzfeldt-Jacob Disease (iCJD) Varyant Creutzfeldt-Jacob Disease (vCJD) Kuru.")
6
İnkübasyon süresi çok uzun
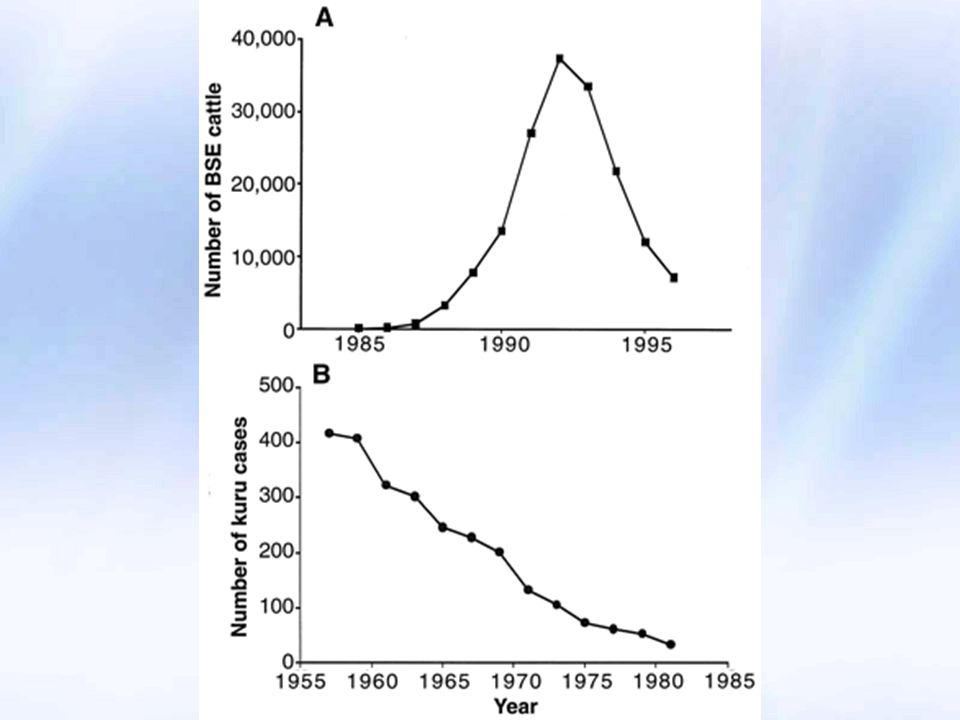
KURU Papua Yeni Gine - Fore halkında bazı ailelerde 1900 başlarında ortaya çıkan arasında 1,100 ölüm Ölenlerin çoğunluğu Kadın Çocuk Yaşlılar İnkübasyon süresi çok uzun An unknown disease appeared in New Guinea in the early 1900’s. It occurred in an isolated group known as the South Fore consisting of about 8,000 people. This group practiced a ritualistic form of cannibalism. When a member of the tribe died, the women and children ate the person's brain and the men ate the muscle tissue. The practice was done to take in the spirit of their ancestors and to ingest a rich source of protein which was difficult to get in the area. It is through these acts of cannibalism that Kuru is believed to have been transmitted among the people. The disease has since been defined as a TSE. Women and children were affected at a rate of about 5 times that of men because they ate the most infectious parts of the body. Between 1957 and 1968, over 1,100 of the South Fore died from kuru. Kuru has essentially disappeared since the termination of cannibalism in the 1970’s. Sporadic cases of kuru still occur, demonstrating that the incubation period is greater than 30 years. The photo depicts Papuans from the central highlands of Papua New Guinea wearing traditional dress and decoration, which is usually reserved for ceremonial or festive occasions. Source: C. Seghers/Photo Researchers, Inc. "Papuans of Papua New Guinea," Microsoft® Encarta® Encyclopedia © Microsoft Corporation.

7
KURU Virolog Carleton Gajdusek (1957)
Çevresel, etken ve toksin ??? Genetik bir hastalık??? Veteriner Dr. William Hadlow (1959) Koyunların scrapie hastalığı ile benzerlik!! Kuru’nun şempanzelere aktarımı ile ilgili deneysel çalışmalar CJD ile benzerliğinin fark edilmesi C Gajdusek Nobel ödülü 1976
 Koyunların scrapie hastalığı ile benzerlik!! Kuru’nun şempanzelere aktarımı ile ilgili deneysel çalışmalar. CJD ile benzerliğinin fark edilmesi. C Gajdusek. Nobel ödülü")
8
KURU Kas güçsüzlüğü Koordinasyon kaybı Gülme krizleri
İmmün yanıt gelişmemesi Beyin dokusunda hasar

9
KURU Kanibalizm ve cenaze ritüellerine bağlı olarak gelişir
Ölen yakınlarının kas dokusu erkekler, beyin ise kadın ve çocuklar tarafından yenir Son enfekte olan kişiler yaşlarındadır

10
CHRONIC WASTING DISEASE
FATAL FAMILIAL INSOMNIA (İNSAN) PrP GEN MUTASYONU 36-60 YAŞ ARASINDA UYKU BOZUKLUĞU KOMA ÖLÜM CREUTZFELDT-JACOB HASTALIĞI SCRAPIE BOVINE SPONGIFORM ENCEPHALOPATHY (BSE) CHRONIC WASTING DISEASE KURU
 PrP GEN MUTASYONU YAŞ ARASINDA. UYKU BOZUKLUĞU. KOMA. ÖLÜM. CREUTZFELDT-JACOB. HASTALIĞI. SCRAPIE. BOVINE SPONGIFORM. ENCEPHALOPATHY (BSE) CHRONIC WASTING DISEASE. KURU.")
11
YAVAŞ VİRUS ENFEKSİYONLARI
Uzun inkübasyon dönemi Çok uzun süre semptom olmaması Genellikle yavaş ilerleyen lethal hastalıklar

12
CANLILAR EUKARYOTE PROKARYOTE NONSELLÜLER VİRUS PRİON HAYVAN
EUBACTERIA BİTKİ ARCHAEOBACTERIA FUNGUS PROTOZOA

13
MİKROORGANİZMALAR ÖKARYOT NONSELLÜLER PROKARYOT PROTOZOA FUNGUS PRİON VİRUS ÖBAKTERİ ARKEBAKTERİ

14
PRİON O halde genom yapılarının özelliği nedir???
Enfeksiyöz ajanlar; çoğalabilmek, nesillerini sürdürebilmek için DNA ve/veya RNA içerir Prionlar; protein yapısındadır ve DNA ve/veya RNA içermediği halde hücre içinde çoğalabilir O halde genom yapılarının özelliği nedir??? 1997 Nobel Fizyoloji-Tıp Dr. Stanley B. Prusiner Scientific American 1984;251:48-57

15
PRION Small proteinaceous infectious particles
Nükleik asitleri inaktive eden yöntemlerden etkilenmez spongiform encephalopathies olarak anılmasının nedeni beyin korteksi ve serebellumda geniş vakuoller oluşturmasıdır The incidence of sporadic CJD is about 1 per million per year. GSS occurs at about 2% of the rate of CJD. Kuru is the condition which first brought prion diseases to prominence in the 1950s. Found in geographically isolated tribes in the Fore highlands of New Guinea. Established that ingesting brain tissue of dead relatives for religious reasons was likely to be the route of transmission. They ground up the brain into a pale grey soup, heated it and ate it. Clinically, the disease resembles CJD. Other tribes in the vicinity with same religious habit did not develop the disease. It is speculated that at some point in the past a tribe member developed CJD, and as brain tissue is highly infectious this allowed the disease to spread. Afflicted tribes were encouraged not to ingest brain tissue and the incidence of disease rapidly declined and is now almost unknown. Prions are proteins that are found in the brains of both normal and afflicted individuals. Prion proteins are found in two forms, the wild type form (PrPc) and the mutant form (PrPsc). Even after much research the actual function of these proteins still remains a mystery. Yet, prions (proteinaceous infectious particles), have been linked to many diseases. The origins of these diseases were previously an enigma; the sources could be linked to neither a bacterial agent nor a viral agent. Via some creative thinking by a man named Stanley B. Prusiner, the prion idea was hypothesized.
 and the mutant form (PrPsc). Even after much research the actual function of these proteins still remains a mystery. Yet, prions (proteinaceous infectious particles), have been linked to many diseases. The origins of these diseases were previously an enigma; the sources could be linked to neither a bacterial agent nor a viral agent. Via some creative thinking by a man named Stanley B. Prusiner, the prion idea was hypothesized.")
16
PRİON Genetik insanda belirlendi Bulaşıcılık Scrapie hayvan modelleri
hipofiz kaynaklı hormon İngiltere’de koyunlarda BSE

17
PRİON Nükleik asit içermez Büyüklüğü 15-40nm.
Hidrofobik yapıda oldukları için kolay agrege olurlar, bu nedenle büyüklük ve dansite yönünden farklılık gösterirler Nukleazlar, UV gibi mikroorganizmalara etkin olan yöntemlerden etkilenmezler Proteinaz K, tripsin gibi maddelerin yüksek dozları etkilidir

18
PRİON Normal kişilerde bulunan PrPc proteininin anormal bir izoformu
PrPsc kDa protein - enfektiviteden sorumlu protein Hasta kişilerin beyin dokularında hastalığın evresi ile orantılı miktarda saptanır Prusiner (1980’ler)
")
19
PRION PROTEİNLERİNİN AGREGE OLACAK FORMA DÖNÜŞÜMÜ
PrPC PrPSc PrPC – PrP hücresel izoformu PrPSC – scrapie benzer izoformu

20
a b Prion Proteinleri (PrPsens PrPres ) PrPsens - PrPc
PrPsens is composed mainly of alpha helicies The conformational change that happens to make the disease protein (PrPres) causes the formation of many beta sheets. This protein becomes resistant to Proteinase K. Thus when the sample is mushed up (homogenised) in the homogeniser (the picture that looks like a centrifuge-but isn’t) and is then digested with proteinase K, all the normal prion protein is digested away (along with most of the other brain proteins) leaving only the resistant protein. The ELISA then tests for the presence of the PrP protein (sens or res, it doesn’t distinguish). The property of aggregation in detergent allows concentration of the sample. TSEs include BSE, CWD (chronic wasting disease in deer & elk), Scrapie (sheep & goats) and (v)CJD (Creutzfeldt Jakob’s disease) in humans. December 23, The first positive BSE cow in the US found in Washington state. Since June 2004 to March, the USDA has screened 284,257 BSE samples. Eight labs in the US: Washington, California, Colorado, Texas, Wisconsin, Georgia, New York, NVSL Lab (Ames, IA). Labs process 30 – 970 samples per day. PrPsens - PrPc Proteinase K sensitive Deterjanda erir PrPres – PrPsc - PrPTSE Proteinase K resistant Deterjanda agrege olur
 causes the formation of many beta sheets. This protein becomes resistant to Proteinase K. Thus when the sample is mushed up (homogenised) in the homogeniser (the picture that looks like a centrifuge-but isn’t) and is then digested with proteinase K, all the normal prion protein is digested away (along with most of the other brain proteins) leaving only the resistant protein. The ELISA then tests for the presence of the PrP protein (sens or res, it doesn’t distinguish). The property of aggregation in detergent allows concentration of the sample. TSEs include BSE, CWD (chronic wasting disease in deer & elk), Scrapie (sheep & goats) and (v)CJD (Creutzfeldt Jakob’s disease) in humans. December 23, The first positive BSE cow in the US found in Washington state. Since June 2004 to March, the USDA has screened 284,257 BSE samples. Eight labs in the US: Washington, California, Colorado, Texas, Wisconsin, Georgia, New York, NVSL Lab (Ames, IA). Labs process 30 – 970 samples per day. PrPsens - PrPc. Proteinase K sensitive. Deterjanda erir. PrPres – PrPsc - PrPTSE. Proteinase K resistant. Deterjanda agrege olur.")
21
PRİON PrPc proteini beyin dokusunda diğer dokulara oranla 50 kat daha fazla bulunur Bu durum TSE olgularının patogenezinde neden MSS tutulduğunu açıklamaktadır PrPsc beyin dokusundan ayrıştırmak güçtür

22
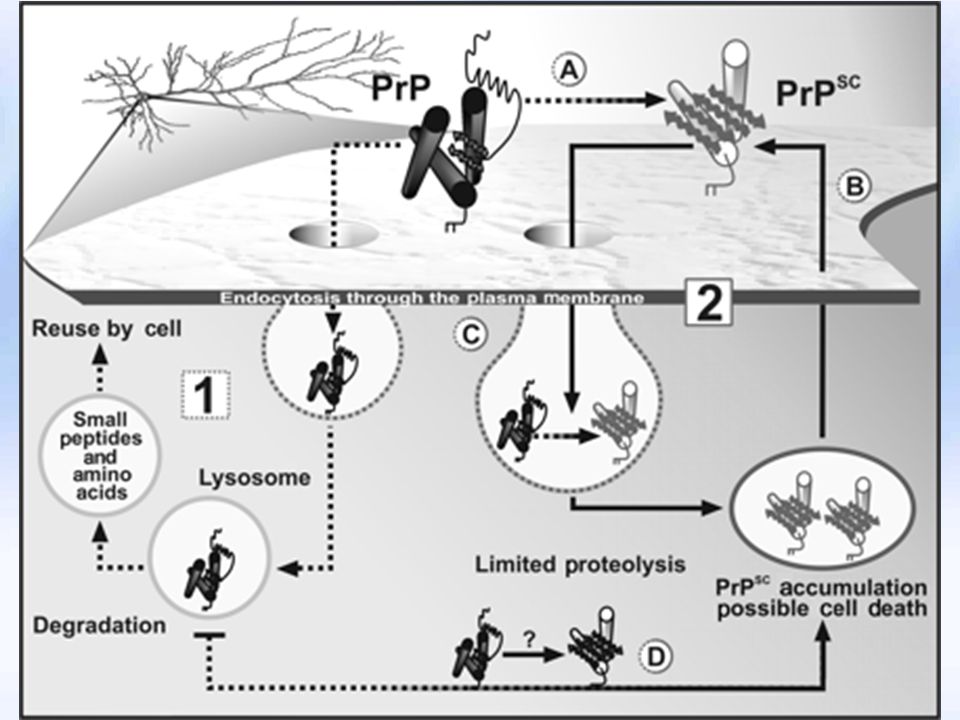
PrPc PrPsc PrPc hücre membranında yarı ömrü 5 saat kadardır
Glikozil fosfatidil inozitol (GPI) aracılığı ile hücre içine girer Endolizozom içine alınır PrPsc dönüşümü muhtemelen hücre içine alım sırasında olmaktadır PrPsc sadece sitoplazmada toplu olarak saptanmaktadır
 aracılığı ile hücre içine girer. Endolizozom içine alınır. PrPsc dönüşümü muhtemelen hücre içine alım sırasında olmaktadır. PrPsc sadece sitoplazmada toplu olarak saptanmaktadır.")
23
GPI

24
bağlayan bir liganttır
PRİON HİPOTEZİ PrP, prP-res’e bağlayan bir liganttır PrP-res PrP-res PrP-res PrP-c PrP-c PrP-c x Protein X bir şaperon olarak PrP-c - prP-res değişimi PrP substrattır PrP-c ve prP-res kompleks oluşturur

25
PrPC PrPSC X-şaperon NMR GÖRÜNTÜLERİ

28
PrP PrP geni (PRNP) 20 kromozom kısa kolu üzerinde yer alır
İnsan PrP proteini 253 AA oluşur N terminalinde 2 hexapepetit ve tekrarlayan oktapepdit yer alır C terminali Glikozil fosfatidil inozitol (GPI) ile ilişkilidir
 ile ilişkilidir.")
29
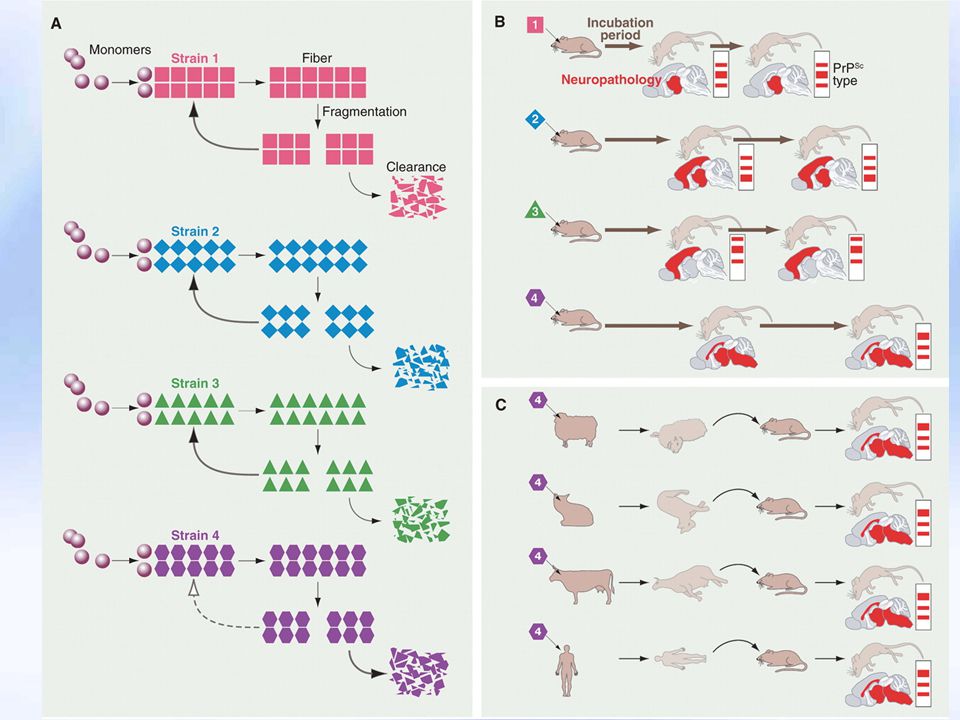
PRİON Viruslarda konak özgüllüğü vardır
Prionlar ise, bulaştıkları türün özelliğini kazanır İlk bulundukları türe ait özelliği kaybeder İnsan Prionu Fare prionu

31
PATOGENEZ - İMMÜN SİSTEM
Dendritik hücreler ve makrofajlar bu ajanların organizmada çoğaldığı ilk hücrelerdir Lenfoid dokularda folliküler dendritik hücrelerde (FDCs) çoğalır ve bu dokuda bulunan sinir hücrelerine invazyon yaparlar
 çoğalır ve bu dokuda bulunan sinir hücrelerine invazyon yaparlar.")
32
TSE ajanının oral yolla alınması
sinir dokusu lenfatik sistem kan perifer beyin ve spinal kord Merkezi sinir sistemi

33
FAE: follikül ilişkili epitel DCs: dendritik hücreler
FDC: folliküler dendritik hücreler GALT:barsak ilişkili lenfoid doku ENS: enterik sinir sistemi Beekes M, McBride P. FEBS Journal 2007, 274:588

34
FDCs – folliküler dendritik hücreler

35
CMGC:çeliak ve mezenterik ganglion kompleksi DRG:dorsal root ganglion
NG:nodos ganglion IML:intermediolateral cell Beekes M, McBride P. FEBSJournal 2007,274:588

36
Beekes M, McBride P. FEBSJournal 2007,274:588

37
İNSANDA vCJD PrPTSE histokimyasal olarak gösterildiği dokular
Üst mezenterik ganglion Sempatik celiak ganglion Dorsal root Trigeminal ganglion Barsak ganglionları Parasempatik ganglion Tonsiller Apendiks YAZARLAR BU BULGULARIN DİKKATLE DEĞERLENDİRİLMESİ GEREKTİĞİNİ VURGULUYORLAR Beekes M, McBride P. FEBSJournal 2007,274:588

38
PATOGENEZ İnkübasyon süresi;
doğal veya deneysel TSE olgularında, klinik semptom olmadan oldukça uzundur Kuru hastalığı ve büyüme hormonuna bağlı gelişen olgularda 40 yıla kadar uzayabilir Hastalık başladıktan sonra remisyon olmadan yavaş olarak ilerler BOS ve kanda enflamasyon bulgusu yoktur Beyin dokusunda herhangi bir enfeksiyöz ajan saptanmaz

39
PATOGENEZ Enfekte kişilerin organ ekstraklarının ultrafiltratlarının enjeksiyonu ile deneysel olarak bulaş sağlanabilir Genetik yapıya da bağlı olarak, enfektivite; Etken Miktar Veriliş yolu ile ilişkilidir İntraserebral enjeksiyon 1 ünit Oral yol ünit

40
PATOGENEZ Etkenin doğal yollarla bulaşı halinde; özellikle BSE ajanı, oral yolda dahil olmak üzere daha fazla enfeksiyözdür Türler arasında bulaş olabilmesi için, alıcı ve verici PrP genlerinin homolog olması gerekir HLA genlerinin de etkisi olmakla beraber, daha azdır

41
PATOGENEZ İnsanda 129. kodonda PrP gen polimorfizmi vardır, Valin veya methionin kodlanabilir Genel populasyonun %50 si homozigottur Sporadik ve iyatrojenik CJD olgularında homozigotluk duyarlılığı arttırmaktadır vCJD olgularının hepsinde methionin/methionin homozigotluğu saptanmıştır

42
Astrositlerin aktivasyonu Mikroglia aktivasyonu
PATOGENEZ PrPsc dönüşümü PrPc PrPsc toplanması PrPsc toplanmasına bağlı sinir ölümü PrPsc ekstrasellüler bölgeye geçmesi Astrositlerin aktivasyonu Mikroglia aktivasyonu Nöron ölümü Gliozis apoptoz

43
PATOGENEZ PrPc, PrPsc ile direkt temastan sonra transforme olur
PrPsc proteolizise dirençli olduğu için enfekte hücre sitoplazmasında toplanır. Sonuçta; nöron ölümü Ekstrasellüler ortama PrPsc çıkışı PrPsc ile diğer nöronlar ya enfekte olur yada apoptoz gelişir PrPsc mikroglia hücrelerini aktive eder ve nörotoksik metaboliter açığa çıkar Astrositlerin aktivasyonu ile gliozis meydana gelir

44
KLİNİK BULGULAR Klinik olarak sessiz dönem ~ 20 yıl
Başlangıç döneminde Hafıza kaybı Koordinasyon bozukluğu, motor kontrol kaybı Psikolojik bozukluklar İlerleyen dönemde Ciddi demans Ataksi, paralizi Ekstrapiramidal ve piramidal sistem ile ilgili bulgular Ensefalit Yaygın nöron kaybı

45
EPİDEMİYOLOJİ İnsan TSE olguları nadirdir
CJD en sık rastlanan formudur Olgular; %85-90 oranında sporadik %10-15 oranında familial <%1 oranda iyatrojenik Epidemiyolojik sürveyansı yapılan ülkelerde de oran benzerdir İnsidans yılda 1-1.7/milyon kişi kadardır

48
GENETİK FORMLAR DAHİL OLMAK ÜZERE BULAŞABİLECEĞİ UNUTULMAMALIDIR
EPİDEMİYOLOJİ İngiltere’de BSE olgularına bağlı gelişen yeni varyant Creutzfeldt-Jacob Disease (vCJD) dışında, hayvanlardan insana geçen olgu saptanmamıştır Özellikle Slovakya ve İsrail’de geçmiş yıllarda saptanmış olguların, gelişen moleküler yöntemler ile kalıtsal olduğu gösterilmiştir ANCAK, İNSAN TSE BÜTÜN FORMLARININ, GENETİK FORMLAR DAHİL OLMAK ÜZERE BULAŞABİLECEĞİ UNUTULMAMALIDIR
 dışında, hayvanlardan insana geçen olgu saptanmamıştır. Özellikle Slovakya ve İsrail’de geçmiş yıllarda saptanmış olguların, gelişen moleküler yöntemler ile kalıtsal olduğu gösterilmiştir. ANCAK, İNSAN TSE. BÜTÜN FORMLARININ, GENETİK FORMLAR DAHİL OLMAK ÜZERE BULAŞABİLECEĞİ UNUTULMAMALIDIR.")
49
RİSK FAKTÖRLERİ Doku grefti Kan transfüzyonu?
Tedavi amacı ile insan, insan kanı, büyük ve küçük baş hayvandan hazırlanan ürünler Hastane sterilizasyon yöntemleri İngiltere’deki olgular; Gıda Tıbbi amaçla büyük baş hayvandan hazırlanan ürünler

50
WHO Scrapie risk faktörlerinin sınıflandırılması
Kategori Enfektivite Doku /vücut sıvısı I Yüksek Beyin, spinal kord II Orta Dalak, tonsil, lenf nodu, ileum, proksimal kolon, plasenta IIIa Bazen Siyatik sinir, hipofiz, adrenal, distal kolon, burun mukozası IIIb Minimal BOS, timus, kemik iliği, karaciğer, akciğer, pankreas, beyaz küre IV Saptanmayan Çizgili kas, kalp, meme, süt, kolostrum, dışkı, böbrek, tiroid, tükrük bezi, over, uterus, testis, seminal sıvı

51
İYATROJENİK CJD ~ inkübasyon süresi Enfeksiyon şekli Hasta sayısı
Beyine giriş yolu ~ inkübasyon süresi Tıbbi uygulama Beyin cerrahisi Steriotaksik EEG 4 2 İntraserebral 20 ay 18 ay Doku nakli Korneal transplant Duramater implant Timpanik transplant 2(+1?) 115 1 Optik sinir Beyin yüzeyi İşitme siniri 33 ay 48 ay Doku ekstraktı Büyüme hormonu Gonadotropin >120 5 Periferik 15 yıl 13 yıl Growth hormones : 132 Human gonadotropines: 4 Meningeal transplant (Lyodura®): 112 Corneal transplant: 3 Invasive procedures (brain electrodes, neurosurgery): 7 Brown et al, Neurology 2000;55: 1 prion positive out of 8318 appendix samples 19 prion positive out of 20 appendices from vCJD autopsy (Hilton DA et al 2002, BMJ 325:633) Spleen and tonsils of vCJD patients are highly infectious in bioassay (Bruce ME et al 2001, Lancet 358:208)
 Optik sinir. Beyin yüzeyi. İşitme siniri. 33 ay. 48 ay. Doku ekstraktı. Büyüme hormonu. Gonadotropin. > Periferik. 15 yıl. 13 yıl. Growth hormones : 132. Human gonadotropines: 4. Meningeal transplant (Lyodura®): 112. Corneal transplant: 3. Invasive procedures (brain electrodes, neurosurgery): 7. Brown et al, Neurology 2000;55: prion positive out of 8318 appendix samples. 19 prion positive out of 20 appendices from vCJD autopsy. (Hilton DA et al 2002, BMJ 325:633) Spleen and tonsils of vCJD patients are highly infectious. in bioassay (Bruce ME et al 2001, Lancet 358:208)")
52
Edinburg – UK CJD istatistikleri
TANIMLANMIŞ veya OLASI CJD NEDENİ İLE ÖLÜM Yıl Şüpheli olgu Sporadik İyatrojenik Familial GSS vCJD Toplam 1990 53 28 5 - 33 1991 75 32 1 3 36 1992 96 45 2 1993 78 37 4 46 1994 118 61 1995 87 35 47 1996 133 40 10 60 1997 162 6 81 1998 154 63 18 89 1999 170 62 15 85 http//

53
TANIMLANMIŞ veya OLASI CJD NEDENİ İLE ÖLÜM
Yıl Şüpheli olgu Sporadik İyatrojenik Familial GSS vCJD Toplam 2000 178 50 1 2 28 82 2001 179 58 4 20 88 2002 163 72 17 94 2003 162 79 5 18 108 2004 114 9 66 2005 124 8 2006 110 65 6 3 80 2007 113 55 67 2008* 2289 958 56 64 32 163ª 1273 *3 mart 2008’e kadar ªNöropatolojik doğrulaması yapılmış olgu sayısı 115

54
Varyant CREUTZFELDT-JAKOB hastalığı
ülke Primer olgu sayısı (yaşayan) Sekonder olgu sayısı: kan transfüzyonu arasında >6ay İngiltere’de yaşamak İngiltere 163(3) 3(0) 166 Fransa 23(2) - 1 İrlanda 4(0) 2 İtalya 1(0) ABD 3*(0) Kanada S Arabistan 1(1) Japonya 1**(0) Hollanda 2(0) Portekiz 2(1) İspanya *S Arabistan’da doğup büyümüş **24 gün ABD yaşamış ( ) http//
 Sekonder olgu sayısı: kan transfüzyonu arasında >6ay İngiltere’de yaşamak. İngiltere. 163(3) 3(0) 166. Fransa. 23(2) - 1. İrlanda. 4(0) 2. İtalya. 1(0) ABD. 3*(0) Kanada. S Arabistan. 1(1) Japonya. 1**(0) Hollanda. 2(0) Portekiz. 2(1) İspanya. *S Arabistan’da doğup büyümüş. **24 gün ABD yaşamış ( ) http//")
55
TIBBİ ÜRÜN GÜVENLİĞİ Donör veya hayvan kaynağının güvenliğinin araştırılması Pürifikasyon aşamaları Doz Son ürün kullanım yeri (intraserebral yol riski en fazla) Olası ise sentetik ürün kullanılmalı
 Olası ise sentetik ürün kullanılmalı.")
56
PRİONLARIN KAN YOLU İLE BULAŞI
TSE ajanı alındıktan sonra, barsak lenfoid sisteminden kan dolaşımına geçmesi olasıdır ancak, deneysel olarak; Keçilerin scrapie Mink TME Kuru vCJD gösterilememiştir

57
PRİONLARIN KAN YOLU İLE BULAŞI
Koyunda doğal yolla veya deneysel scrapie de, klinik öncesi ve klinik dönemde kanda saptanmış Hamsterlerde deneysel scrapie hastalığında düşük düzeyde “viremi?”nin en az 40 gün sürebileceği gösterilmiş İnsan kan donörlerinde de, vCJD klinik öncesi dönemde bulunabilir???

58
KAN VE DOKU DONÖRÜ OLARAK KULLANILMAMALIDIR
TIBBİ ÜRÜN GÜVENLİĞİ Subakut ensefalopatisi olan olgular Ailede familial CJD, GSS, fFI olgusu olan kişiler Geçmişte hipofiz kaynaklı hormon tedavisi almış yada duramater grefti uygulanmış kişiler KAN VE DOKU DONÖRÜ OLARAK KULLANILMAMALIDIR

59
TIBBİ ÜRÜN GÜVENLİĞİ Kanda enfeksiyöz partikül varmıdır??
Kan bankacılığı önlem almalımıdır?? Bazı ülkeler arasında 6 aydan fazla İngiltere’de bulunmuş kişileri donasyonda çıkartıyor Bazı ülkeler lökosit ayrımı yaparak kan kullanıyor İngiltere kaynaklı plazmalardan ürün hazırlanmıyor Lenfoid doku ile ilişkili cerrahi işlemler risklimidir???

60
PRİON DİRENÇLİDİR Filtreler Radyasyon UV (254nm) Kaynatma Nukleaz
Proteaz Divalen katyonlar Asit Radyasyon Kaynatma Kuru ısı Alkol Betapropionolakton Formalin DİRENÇLİDİR

61
PRİON İNAKTİVASYONU Proteazlar ile uzun süreli muamele
Sodyum dodesil sülfat ve üre ile kaynatma 132°C otoklavda 4.5 saat (enfekte doku) Guanidin izotiyosiyanat/ hidroklorid 4.0 M NaOH 1N Na hipoklorid %2 132°C OTOKLAV 4.5 SAAT
 Guanidin izotiyosiyanat/ hidroklorid 4.0 M. NaOH 1N. Na hipoklorid %2. 132°C OTOKLAV. 4.5 SAAT.")
62
TIBBİ GÜVENLİK STERİLİZASYON
ÖN TEMİZLİK; Otoklavda °C – 20 dak (türler arasıda direnç farkı olabilir!!!) Sodyum hidroksit 1-2mol/l – 1 saat - 20 °C Sodyum hipoklorit %2 – 1 saat - 20 °C NORMAL STERİLİZASYON PROSEDÜRÜ Riskli bir hastaya cerrahi işlem yapılacaksa WHO önerileri dikkate alınmalıdır
 Sodyum hidroksit 1-2mol/l – 1 saat - 20 °C. Sodyum hipoklorit %2 – 1 saat - 20 °C. NORMAL STERİLİZASYON PROSEDÜRÜ. Riskli bir hastaya cerrahi işlem yapılacaksa WHO önerileri dikkate alınmalıdır.")
63
İMMÜN YANIT Doğal enfeksiyon veya deneysel koşullarda humoral ve hücresel immün cevapta değişiklik olmaz PrP’ye karşı oluşan bir antikor cevabı yoktur Dolayısıyla, asemptomatik olguların saptanması bağışık yanıt aracılığı ile olası görülmemektedir

64
MİKROBİYOLOJİK TANI Humoral immün cevap oluşmaması nedeniyle, tanıda antikor oluşumunu gösteren kolay bir test bulunmamaktadır Olguların tanısı genellikle postmortem olarak konulmaktadır Prion 15B3 epitopuna karşı geliştirilen monoklonal antikor (mAb) ile tanı olasıdır
 ile tanı olasıdır.")
65
Prion 15B3 epitopu monoklonal antikor (mAb)ile PrPTSE tanısı
15B3 epitope Nature 390 (6655) : 74-77, 1997
 : 74-77,")
66
ELISA TESTİ Immunoassay - prion specific antikor 15B3
Geniş tür spesifikliği İnsan Koyun rodent Hastalık ile ilişkili anormal PrP saptanması Immünpresipitat Western blot FACS Sensitivite 10 – 100 enfeksiyöz ünit/ ml kan

67
Nanomechanical resonator array ve secondary mass labeling
2ng/ml miktarda PrP’yi antemortem olarak saptamayı sağlamaktadır Varsney M et al. Analitical Chemistry 2008

Benzer bir sunumlar