Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
Anabilim Dalı Nöroloji Bilim Dalı Olgu Sunumu 26 Eylül 2014 Cuma Uzm. Dr. Emek Uyur Yalçın

2
ÇOCUK NÖROLOJİSİ BİLİM DALI OLGU SUNUMU
26/09/2014 ÇOCUK NÖROLOJİSİ BİLİM DALI OLGU SUNUMU Dr. Emek Uyur Yalçın

3
Olgu: 15 aylık erkek hasta “Şikayet”
Başını tutamama, desteksiz oturamama. “Öykü” Aile son üç aydır hastanın genel olarak güçsüz olduğunu fark etmiş. Daha önceden başını tutabilirken son zamanlarda başını tutmakta zorlandığını, desteksiz oturamadığını gözlemlemiş.

4
Olgu: “Özgeçmiş” Prenatal takibinde özellik yok.
G2P2A0 anneden, miadında, C/S ile 4100 gram olarak doğmuş. Postnatal adaptasyon sorunu olmamış Başını tutma : 2 ay Desteksiz oturma: 6-7 ay Dil gelişimi: 11 aylıkken 3-4 kelimesi varmış, şu an tek kelimesi var. Yürüme: Yok.

5
Olgu: “Soygeçmiş” Anne: 32 yaşında, sağlıklı
Baba: 36 yaşında, sağlıklı 1. derece kuzen evliliği 12 yaşında sağlıklı kız kardeş

6
Olgu: “Fizik muayene” Bilinci açık, kooperasyonu kısıtlı, huzursuz, göz takibi var, etrafla ilgisi zayıf, ortak dikkati var, yabancıları yadırgaması var. Baş traksiyonda geride kalıyor, yaygın bir tonus artışı mevcut, DTR’leri hiperaktif, bilateral klonus mevcut, ekstremiteleri hareketli ancak kas gücü net değerlendirilemedi. Ses ve dokunmayla irkiliyor, bazen distonik postür alıyor. Diğer sistem muayeneleri doğal. Baş çevresi: (3. p.’nin altı) Boy: 76 cm ( p) Tartı: 9500 g ( p)
 Boy: 76 cm ( p) Tartı: 9500 g ( p)")
7
Olgu ‘’Tetkikler’’ Hemogram, genişletilmiş biyokimya,
Amonyak, laktat, kan gazı Tiroid fonksiyon testleri, B12, folat, homosistein TANDEM-MS/MS, idrar organik asit analizi Normal Göz muayenesi İşitme testi EKO EMG

13
Patolojik bulgular: Mikrosefali Kazanılmış becerilerde kayıp
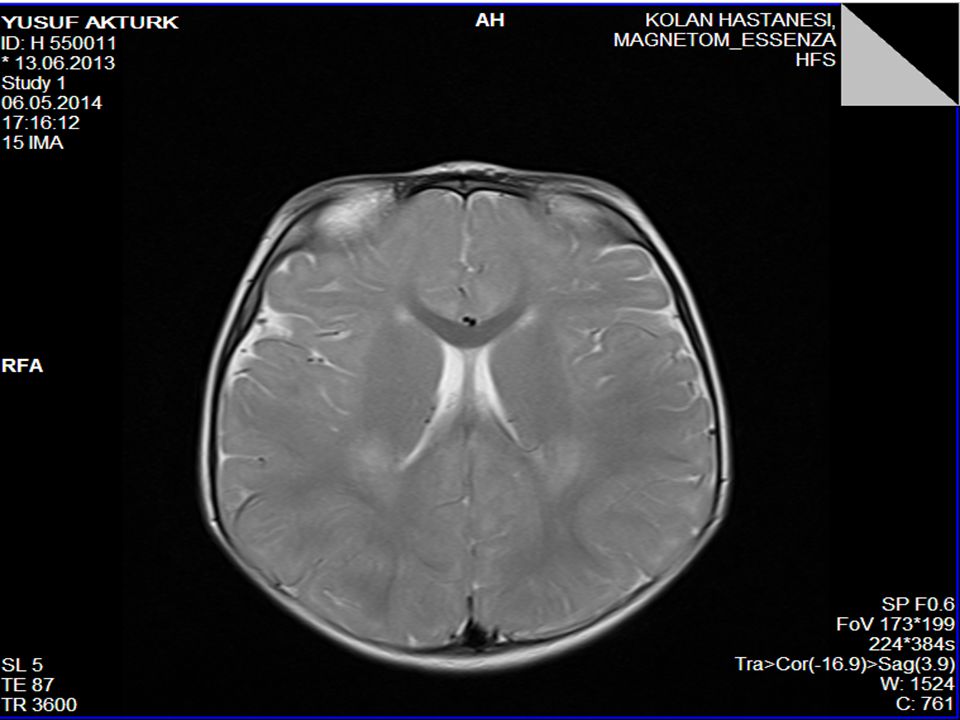
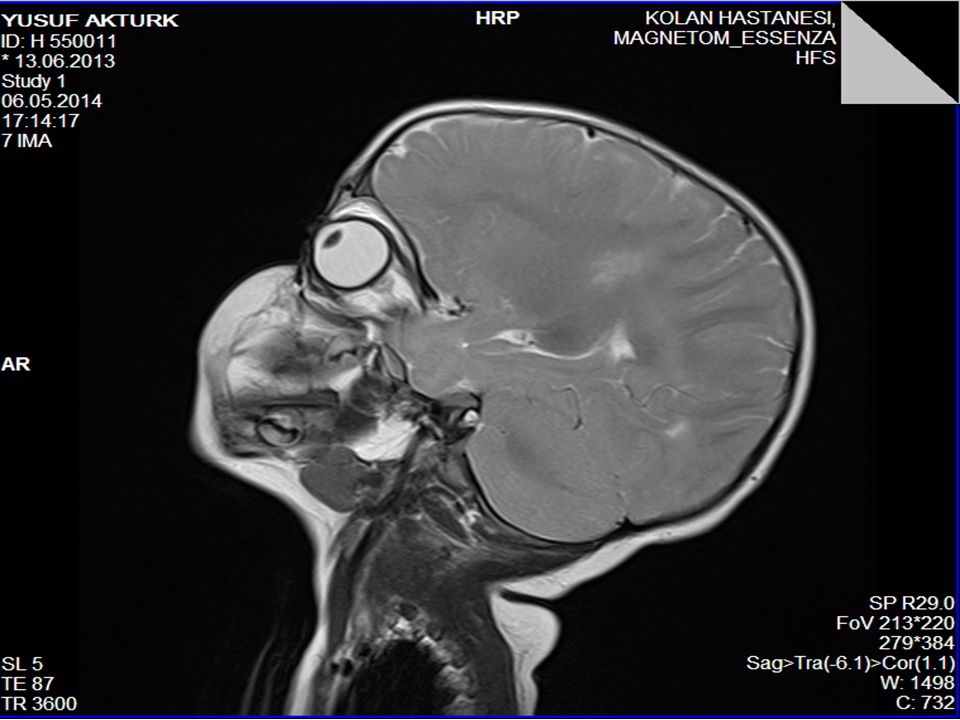
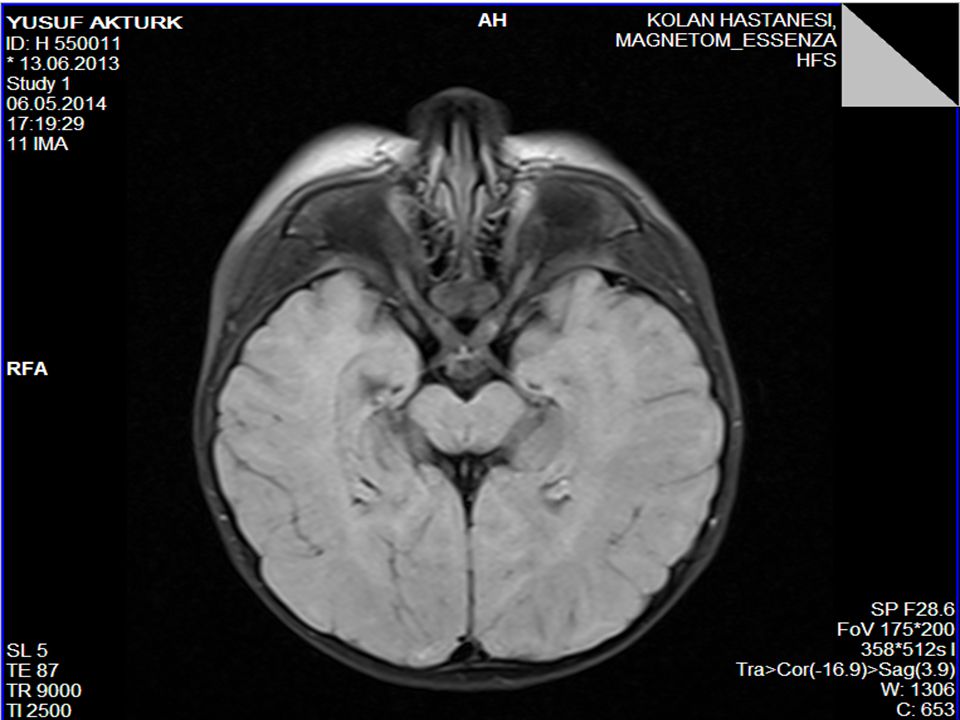
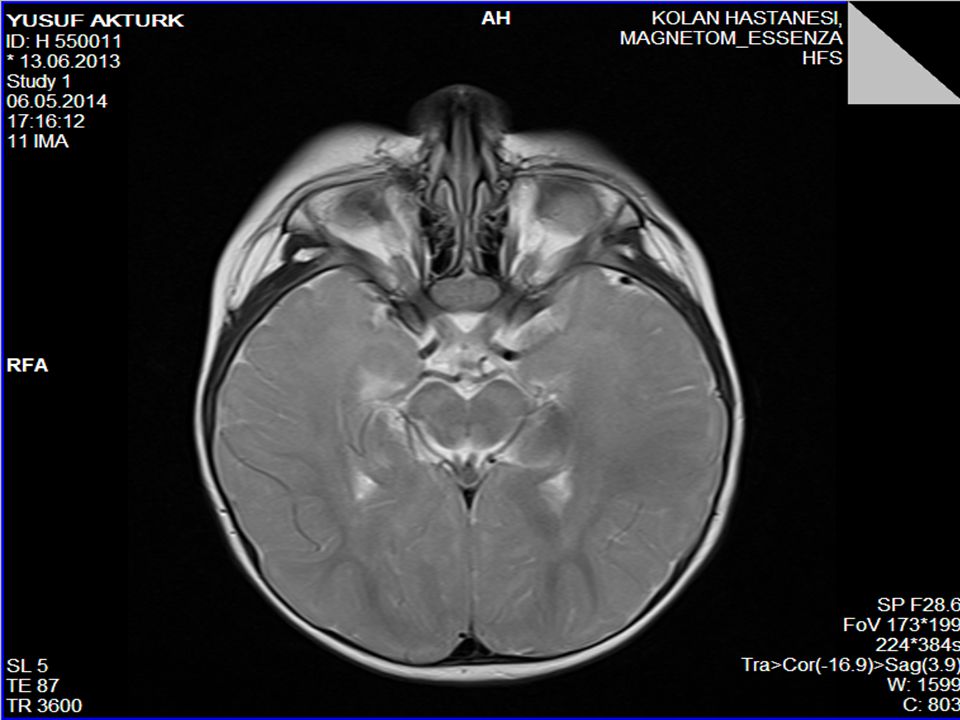
Piramidal bulgular Ses-dokunma ile tetiklenen distonik postür ve ekstansör spazmlar (hiperakuzi) Kranyal MRG’sinde serebral hemisferlerde derin ak maddede simetrik T2 ve FLAİR sekanslarda sinyal değişiklikleri (myelinizasyon defekti) ve optik sinirlerde kalınlaşma 1. derece kuzen evliliği
 Kranyal MRG’sinde serebral hemisferlerde derin ak maddede simetrik T2 ve FLAİR sekanslarda sinyal değişiklikleri (myelinizasyon defekti) ve optik sinirlerde kalınlaşma. 1. derece kuzen evliliği.")
14
ÖN TANI?

15
Ayırıcı Tanı: Nörometabolik hastalıklar

16
Nörometabolik hastalıklar:
Çocukluk çağı nörometabolik hastalıkları özgün bir enzimin eksikliği sonucu biyokimyasal maddelerin sentezlenememesi veya toksik metabolitlerin birikimi sonucu beyin dokusunun hasar görmesi ile karakterizedir. Etyolojisi açıklanamayan santral sinir sistemine ait belirtilerle başvuran çocuklarda ayırıcı tanıda düşünülmelidir. Klinik belirtilerin başlama yaşı, ortaya çıkış şekli ve seyri tanı konulmasında önemlidir.

17
Nörometabolik hastalıklar:
Nörometabolik hastalıklar gri cevher, beyaz cevher veya her ikisini birden etkileyebilir. Hastalıklar; patogenez, tutulan organel ve beyin tutulum lokalizasyonlarına göre sınıflandırılmaktadır. Görüntüleme bulguları genellikle nonspesifik olmasına rağmen, tutulum alanlarına göre ayırıcı tanı yapmak mümkün olabilmektedir.

18
a)Sadece beyaz cevher tutulumu
* Erken dönemde derin beyaz cevher 1- Krabbe hastalığı 2- Peroksizomal hastalıklar 3- Metakromatik lökodistrofi 4- Fenil ketonüri 5- Maple syrup urine hastalığı 6- Lowe sendromu 7- Hiperhomosistinemi * Erken dönemde subkortikal beyaz cevher 1- Fibrinoid lökodistrofi (Alexander hastalığı) 2- van der Knaap hastalığı (vakuoler megalensefali) 3- Galaktozemi 4- L2 –hidroksi glutarik asiduri * Myelinizasyon yokluğu 1- Pelizaeus-Merzbacher 2- Trikotiodistrofi 3- 18q sendromu * Nonspesifik beyaz cevher tutulumu 1- Üre siklusu bozuklukları3 2- Nonketotik hiperglisinemi 3- Herhangi bir beyaz cevher hastalığının son evresi
 2- van der Knaap hastalığı. (vakuoler megalensefali) 3- Galaktozemi. 4- L2 –hidroksi glutarik asiduri. * Myelinizasyon yokluğu. 1- Pelizaeus-Merzbacher. 2- Trikotiodistrofi q sendromu. * Nonspesifik beyaz cevher tutulumu. 1- Üre siklusu bozuklukları3. 2- Nonketotik hiperglisinemi. 3- Herhangi bir beyaz cevher hastalığının son evresi.")
19
b) Sadece gri cevher tutulumu
* Kortikal gri cevher 1- Nöronal seroid lipofuksinozis 2- Mukolipidozis Derin gri cevher 1- Leigh hastalığı 2- Juvenil Huntington hastalığı 3- MELAS 4- Hallervorden-Spatz hastalığı

20
c) Beyaz ve gri cevherin birlikte tutulumu
* Derin gri cevher tutulumu 1- Krabbe hastalığı 2- GM1, GM2 gangliozidozlar 3- Canavan hastalığı 4- Akçaağaç şurubu hastalığı 5- Kearns-Sayre sendromu 6- MELAS 7- Leigh sendromu 8- Wilson sendromu 9- Cockayne sendromu * Sadece kortikal gri cevher 1- ALPER hastalığı 2- Menkes hastalığı 3- Mukopolisakkaridozlar 4- Lipid depo hastalıkları 5- Peroksizomal hastalıklar 6- Konjenital musküler distrofiler

21
Lizozomal depo hastalıkları
Metakromatik lökodistrofi GM1 Gangliozidoz GM2 Gangliyozidoz (Tay-Sachs, Sandhoff) Niemann-Pick C hastalığı Gaucher tip 2/3 Krabbe
 Niemann-Pick C hastalığı. Gaucher tip 2/3. Krabbe.")
22
Lökosit içi lizozomal enzim aktivitesi (galaktoserebrosidaz): (nmol/17 saat/mg protein)
0.44 (107.3+/-35.8)
")
23
Krabbe Hastalığı “Globoid hücreli lökodistrofi”
OR kalıtılan lizozomal depo hastalığı ‘Galaktoserebrosidaz’ (GALC) enzim eksikliği Bu enzim; ak madde myelinizasyonu sırasında lipozomal hidroliz ve galaktolipid formülasyonunda rol almaktadır. GALC geni, 14q31. kromozomunda lokalize 70’den fazla mutasyon tanımlanmış.
 enzim eksikliği. Bu enzim; ak madde myelinizasyonu sırasında lipozomal hidroliz ve galaktolipid formülasyonunda rol almaktadır. GALC geni, 14q31. kromozomunda lokalize. 70’den fazla mutasyon tanımlanmış.")
24
Klinik özellikler: İnfantil Tip:
Semptomlar sıklıkla 2-5 ay arasında ortaya çıkmaktadır. İrritabilite, nöromotor gelişimde duraklama veya gerileme, ekstremitelerde spastisite, aksiyel hipotoni, arefleksi, optik atrofi, mikrosefali, ışık-ses-dokunmayla ortaya çıkan tonik ekstansör spazmlar. Hızla deserebre postür gelişmekte ve çoğu olgu 2 yaş civarında kaybedilmektedir.

25
Klinik özellikler: Juvenil Tip:
Belirtilerin başlangıcı 2-6 yaş arasındadır. Kazanılmış becerilerin kaybı ve görme kaybı bu olgularda daha ön planda. Bu tipin erken başlangıçlı tipten ayırt ettirici özelliği, periferik nöropatinin olmamasıdır. Geç infantil ve juvenil tip tanısı alan olgular, hızla persistan vejetatif duruma ilerlemekte ve tanıdan sonraki 2-7 yıl içerisinde kaybedilmektedir.

26
Klinik özellikler: Erişkin Tip:
Semptomlar adolesan dönemde veya erişkin dönemde ortaya çıkmaktadır. El becerisinin kaybı, ekstremitelerde parestezi, güçsüzlük, nöropati, kaslarda atrofi ve skolyoz ön planda. Bazı olgularda kognitif fonksiyonlar korunmaktadır.

27
Tanı: Kranyal MRG’sinde;
İnfantil başlangıçlı tipte; serebral derin ak madde (periventriküler, sentrum semiovale), dentat nükleus, serebellar ak maddeyi içeren yaygın demyelinizasyon. Juvenil ve erişkin başlangıçlı tipte; ek olarak diffüz atrofi, parieto-oksipital bölgelerde ve/veya kortikospinal traktta T2 sinyal artışı
, dentat nükleus, serebellar ak maddeyi içeren yaygın demyelinizasyon. Juvenil ve erişkin başlangıçlı tipte; ek olarak diffüz atrofi, parieto-oksipital bölgelerde ve/veya kortikospinal traktta T2 sinyal artışı.")
28
Tanı: Sinir-ileti çalışmaları:
İnfantil başlangıçlı olgularda, motor ve duysal sinir ileti hızlarında belirgin yavaşlama saptanabilmekte (presemptomatik dönemde olsalar bile). Daha geç başlangıçlı olguların ise %20’sinde sinir-ileti çalışmalarında yavaşlama tespit edilmiş. Sinir-ileti çalışmalarındaki patolojik bulguların ağırlığı ile hastalığın ciddiyeti arasında bir korelasyon söz konusu. Siddigi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology 2006; 67:263
. Daha geç başlangıçlı olguların ise %20’sinde sinir-ileti çalışmalarında yavaşlama tespit edilmiş. Sinir-ileti çalışmalarındaki patolojik bulguların ağırlığı ile hastalığın ciddiyeti arasında bir korelasyon söz konusu. Siddigi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology 2006; 67:263.")
29
Tanı: Semptomatik çocukların VEP, BAEP ve EEG incelemelerinde de patolojik sonuçlar elde edilmiş. BOS incelemesinde; proteinin artışı mevcut, ancak tanı için rutinde yapılmamaktadır. 25 semptomatik infantil Krabbe olgusuna lomber ponksiyon yapılmış, 23’ünde (%92’sinde) BOS proteinin artmış olduğu gösterilmiş. Duffner PK, Barczykowski A, Jalal K, et al. Early infantile Krabbe disease: results of the Worl-Wide Krabbe Registry. Pediatr Neurol 2011; 45:141
 BOS proteinin artmış olduğu gösterilmiş. Duffner PK, Barczykowski A, Jalal K, et al. Early infantile Krabbe disease: results of the Worl-Wide Krabbe Registry. Pediatr Neurol 2011; 45:141.")
30
Tanı: Patolojik inceleme:
Periferik sinir demyelinizasyonu ve PAS pozitif boyanan multinükleer globoid hücreler Erken başlangıçlı formda hipomyelinizasyon, geç başlangıçlı formda segmental demyelinizasyon ön planda.

31
Tanı: Kesin tanı; periferik kan lökositlerinde veya fibroblast kültüründe galaktoserebrosidaz enzim aktivitesinin ölçümü ile konur. Rezidüel enzim aktivitesi; klinip tip veya prognoz için bir gösterge niteliği taşımamaktadır. Moleküler genetik çalışma (bilinen mutasyonlarda prenatal danışma verilebilir)
")
32
Tandem mass spektrometri yöntemini kullanarak, YD döneminde kuru kan örneğinde direkt galaktoserebrozidaz enzim aktivitesine bakılmış (New York). Ancak yararlılığı sınırlı, çünkü ne enzim aktivitesinin ölçülmesi ne de genetik mutasyonun saptanması fenotip hakkında öngörü sağlamamakta. Duffner PK, Caggana M, Orsini JJ, et al. Newborn screening for Krabbe disease: the New York state model. Pediatr Neurol 2009;

33
Tedavi: Semptomatik hastalarda irritabilite ve spastisiteye yönelik destekleyici tedavi söz konusu. Presemptomatik dönemde infantil ve juvenil Krabbe olgularında kök hücre transplantasyonunun yararlı olabileceği bildirilmekte; ancak bu uygulamanın uzun dönem etkileri bilinmiyor.

34
Tedavi: İnfantil Krabbe tanısı alan; 11 asemptomatik (12-44 gün) ve 14 semptomatik ( gün) olguya transplantasyon yapılmış. 3 yıllık izlemde; 11 asemptomatik olgunun 10’unda dil gelişimi yaşına uygun olarak değerlendirilmiş, 9’unda yaşına uygun kognitif gelişim saptanmış, 4’ünde hafif-orta motor gerilik tespit edilmiş. 14 semptomatik olgunun ancak 6’sı yaşamış, ölüm oranının nakil yapılmamış kontrol grubuna göre istatiksel olarak anlamlı bir farklılık göstermediği gözlenmiş. Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umblical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med 2005; 352:2069

35
Tedavi: İlk kanıtlar kök hücre transplantasyonunun hayatın ilk bir ayında (presemptomatik dönemde) uygulandığında faydalı olabileceğini göstermiş olup, çalışma sayısı oldukça yetersiz.
 uygulandığında faydalı olabileceğini göstermiş olup, çalışma sayısı oldukça yetersiz.")
36
Krabbe hastalığını düşün!
Son Söz: Kazanılmış becerilerde kayıp, mikrosefali, ses-dokunma ve ışık gibi uyaranlara ekstansör spazm şeklinde yanıt (hiperakuzi) bulguları olan olgularda, Kranyal MRG’sinde ak madde tutulumu ve optik sinirde kalınlaşma saptanırsa, Krabbe hastalığını düşün!
 bulguları olan olgularda, Kranyal MRG’sinde ak madde tutulumu ve optik sinirde kalınlaşma saptanırsa, Krabbe hastalığını düşün!")
37
Bir gökdelen tepesinde öğle yemeği; Rockefeller Merkezi , John C Ebbets, 1932

Benzer bir sunumlar









>")










