Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Mitokondriyal hastalıklar

2
Mitokondriyal genetik Mitokondriyal hastalıklar
Tarihçe Mitokondri Mitokondriyal genetik Mitokondriyal hastalıklar mtDNA nDNA Tanı Neredeyiz?

3
Tarihçe Luft R., Ikkos D., Palmieri G., A case of severe hypermetabolism of non-thyroid origin with a defect in the maintenance of mitochndrial respiratory control: a correlated clinical, biochemical and morphological study. J. Clin. Invest., 41, , 1962. Kas dokusunda morfolojik olarak anormal mitokondri Biyokimyasal olarak oksidasyon ve fosforilasyonun bozukluğu Biyokimyasal defekt ile klinik özellikler arasında uyum 1960 – mitokondriyal genlerdeki defektler miyopatilerde rol oynayabilir. 1963 – W. King Engel, Modifiye gomori trikrom boya Ragged red fibers 1988 – Mitokondriyal hastalıkların moleküler çağı başlar. – nDNA ve mitokondriyal hastalıklar Bugün –

5
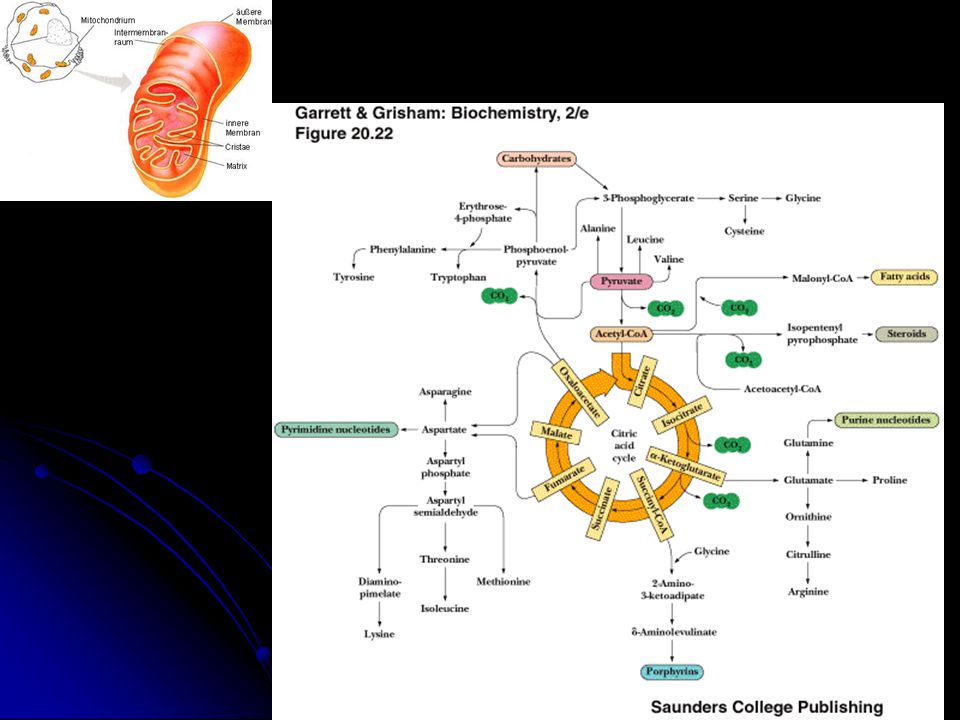
Piruvat dehidrogenaz kompleksi (PDHC)
Carnitine siklusu β-oksidasyon sistemi Krebs siklusu

6
ETZ + Oksidatif fosforilasyon= Solunum zinciri (OXPHOS)
")
7
İki genom tarafından kontrol edilir
OXPHOS İki genom tarafından kontrol edilir mtDNA nDNA

8
Kompleks II, koenzim Q10 ve sitokrom C sadece nDNA ile kodlanır.
Solunum zincirini oluşturan ~80 proteinin 13’ ü mitokondriyal genom tarafından kalanlar ise nüklear genom tarafından kodlanır. 7 gen – komplex I 1 gen (sitokrom b) – kompleks III 3 gen – kompleks IV 2 gen – kompleks V (ATP sentaz) Kompleks II, koenzim Q10 ve sitokrom C sadece nDNA ile kodlanır. mtDNA: 2 rRNA ve 22 tRNA (toplam 37 gen)
 – kompleks III. 3 gen – kompleks IV. 2 gen – kompleks V (ATP sentaz) Kompleks II, koenzim Q10 ve sitokrom C sadece nDNA ile kodlanır. mtDNA: 2 rRNA ve 22 tRNA (toplam 37 gen)")
9
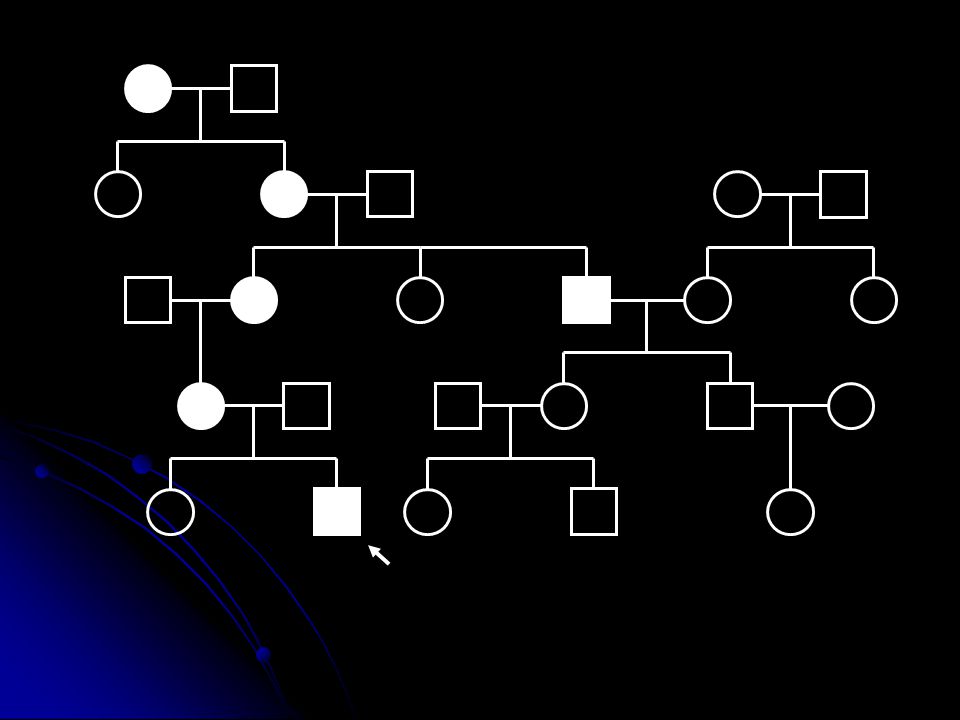
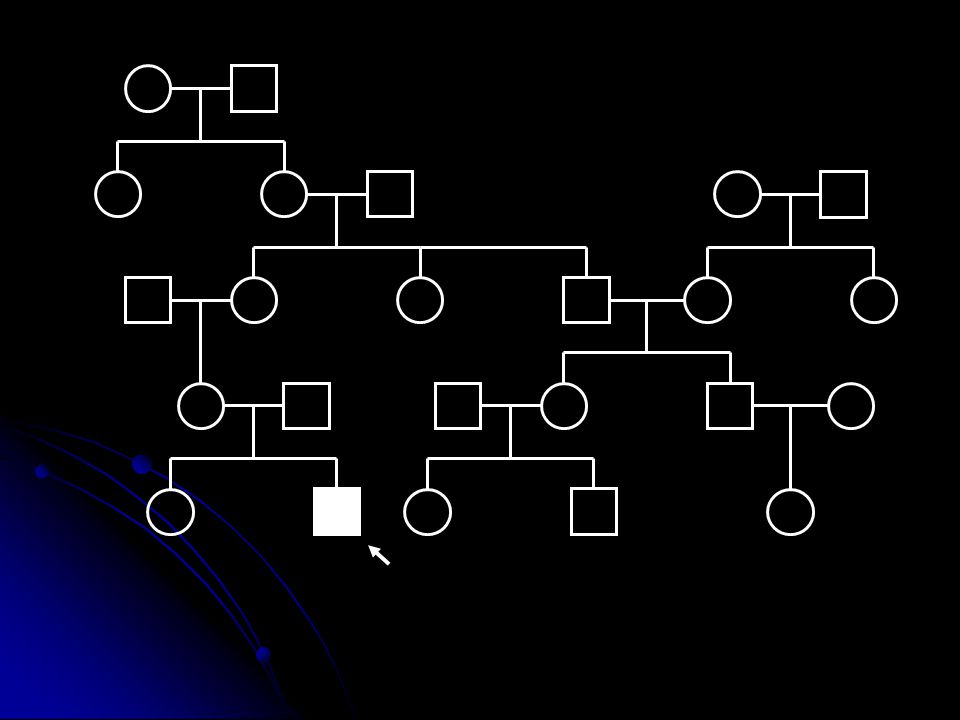
Mitokondriyal genetiğin özellikleri
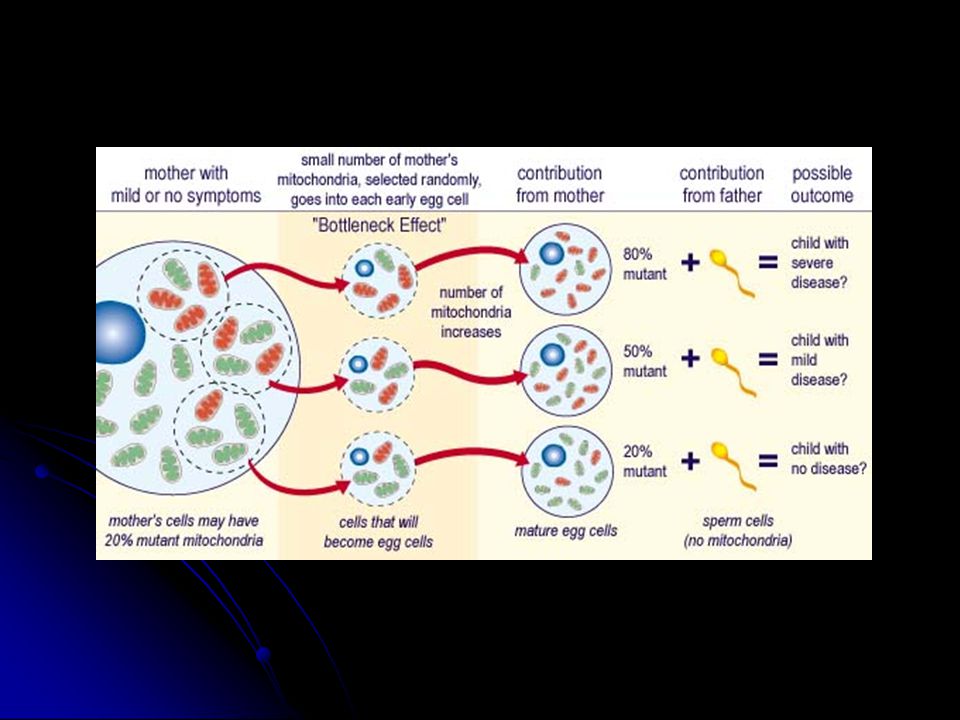
Heteroplazmi ve eşik etkisi Genellikle mutant DNA ve yabanıl tip DNA bir arada bulunur Hücredeki mutant/yabani oranı oksidatif fosforilasyonun o hücre için ne kadar etkileneceğini belirler. (Mutant yük). Enerji ihtiyacı o hücre için mutant DNA eşik değerini belirler (threshold effect).
. Enerji ihtiyacı o hücre için mutant DNA eşik değerini belirler (threshold effect).")
10
Mitokondriyal genetiğin özellikleri
Mitotik segregasyon Hücre bölünmesinde mutant DNA oranı yavru hücrelerde değişebilir. Klinik fenotipin ilerleyen yaşla birlikte değişimi Maternal kalıtım Mendel kalıtım kurallarından farklı bir kalıtım şekli

14
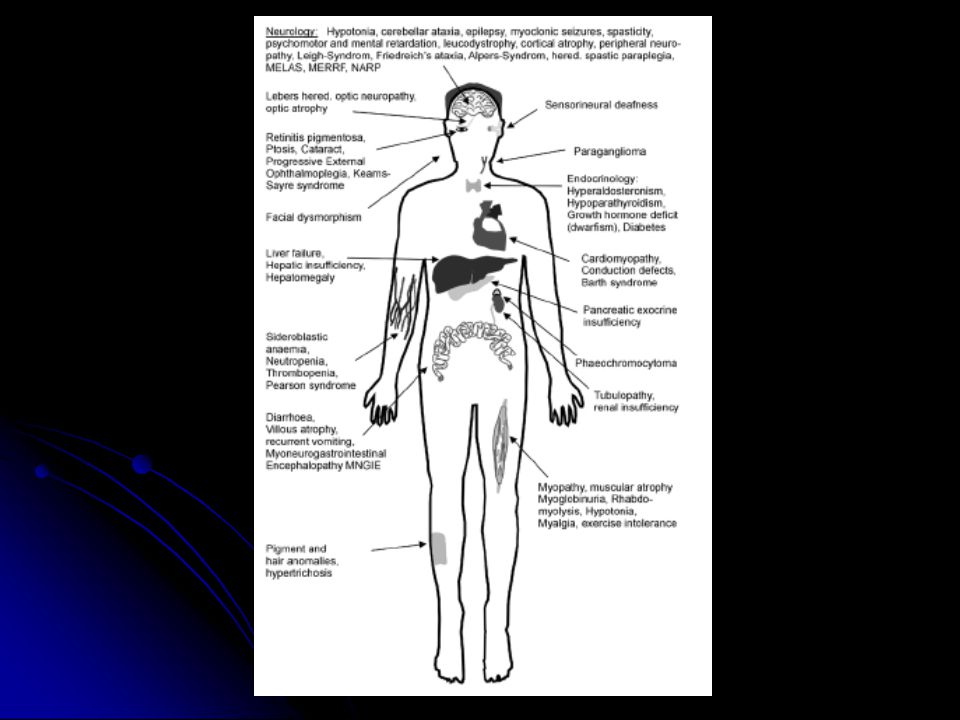
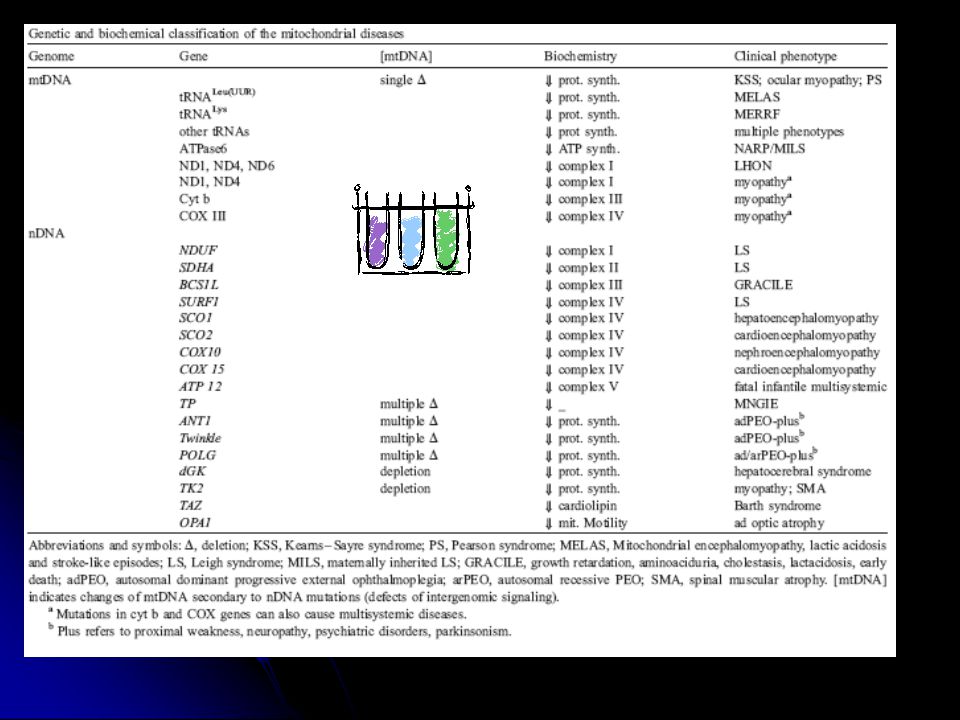
Mitokondriyal hastalıklar
mtDNA hasarı ile ilişkili hastalıklar nDNA hasarı ile ilişkili hastalıklar mtDNA ve nDNA hasarı ile ilişkili

15
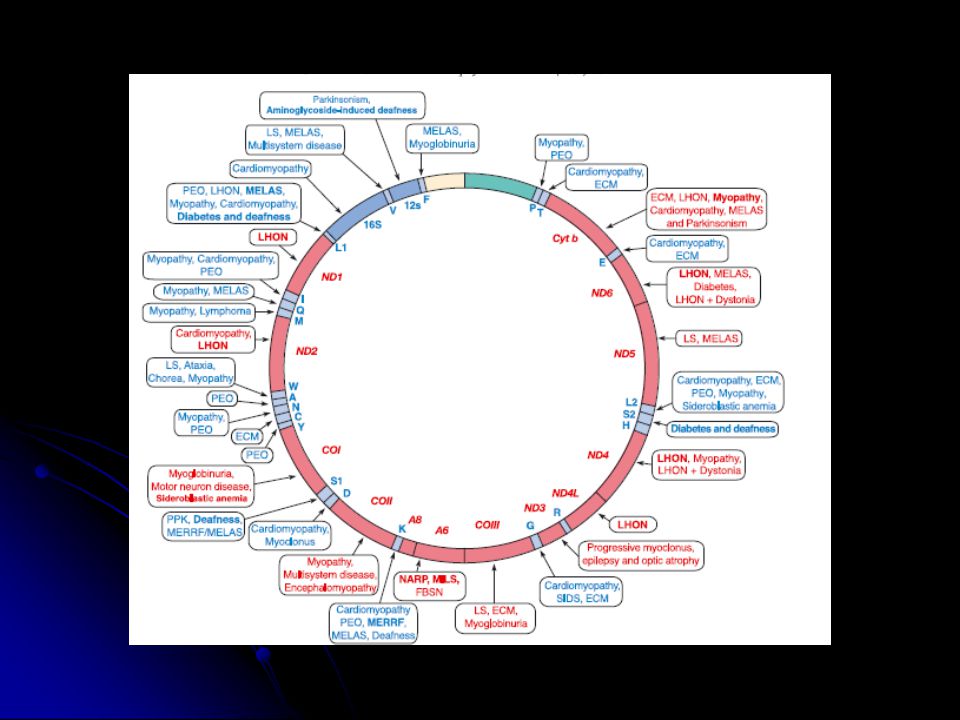
mtDNA hasarı Protein sentezi ile ilgili genlerdeki mutasyonlar
tRNA ve rRNA genleri Protein kodlayan genlerdeki mutasyonlar

17
Mitokondriyal protein sentezi bozuklukları
Mitokondriyal DNA delesyonları Pearson sendromu (PS): sideroblastik anemi ve ekzokrin pankreas bozukluğu ile görülür. Delesyon spektrumu geniş. Kearns-Sayre sendromu (KSS): progresif eksternal oftalmopleji, pigmenter retinopati, ataxia, demans laktik asidozis ile seyreder. Delesyon spektrumu geniş.
: sideroblastik anemi ve ekzokrin pankreas bozukluğu ile görülür. Delesyon spektrumu geniş. Kearns-Sayre sendromu (KSS): progresif eksternal oftalmopleji, pigmenter retinopati, ataxia, demans laktik asidozis ile seyreder. Delesyon spektrumu geniş.")
18
Mitokondriyal protein sentezi bozuklukları
Sporadik PEO Progresif eksternal oftalmopleji: Göz küresi dışındaki kasların felci ile göz hareketlerinin bozulması Bazen proksimal ekstremite zayıflığı mtDNA delesyonu (özellikle 4.9 kb CD) ve bazen duplikasyonlar Maternal kalıtım izlenebilir.
 ve bazen duplikasyonlar. Maternal kalıtım izlenebilir.")
19
Mitokondriyal protein sentezi bozuklukları
Mitokondriyal DNA duplikasyonları KSS, diabetes mellitus ve sağırlık gösteren hastalarda tek delesyonlara ek olarak duplikasyon içeren mtDNA molekülleride bulunabilir. (Associated phenotypes). Nadirdir ve genellikle maternal kalıtımlıdır.
. Nadirdir ve genellikle maternal kalıtımlıdır.")
20
Mitokondriyal protein sentezi bozuklukları
Nokta mutasyonları Çok sayıda nokta mutasyonu tanımlanmıştır. Multisistemik veya doku spesifik olabilir. De novo veya kalıtılmış olabilir.

21
Mitokondriyal protein sentezi bozuklukları
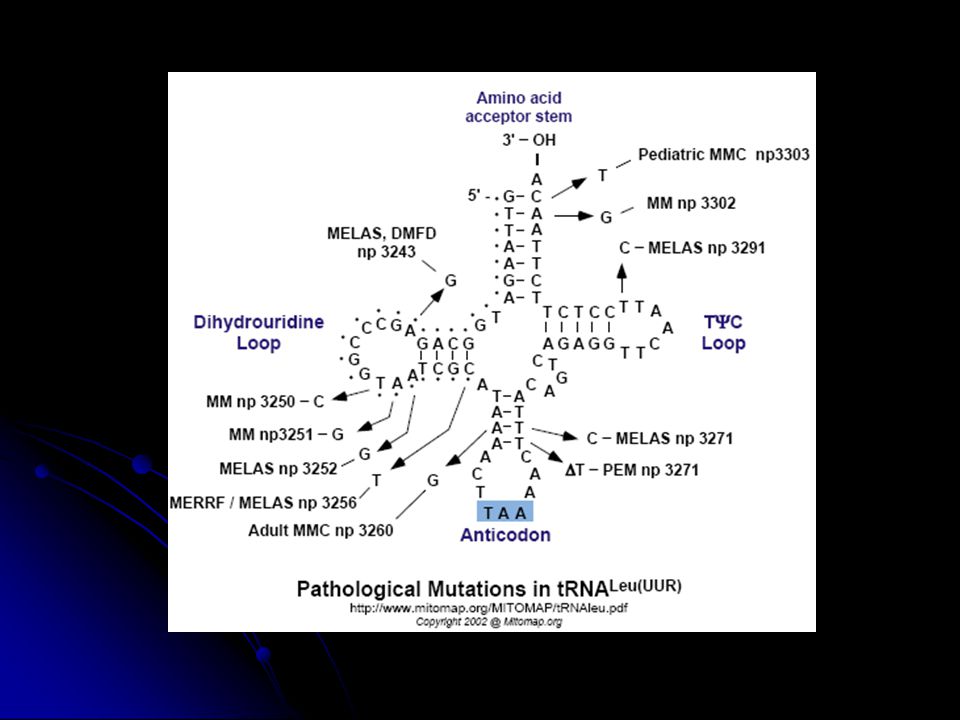
MELAS: mitokondriyal ensefalomiyopati, laktik asidosiz, nöbet benzeri episodlar Migren benzeri baş ağrısı, nöbetlere bağlı kortikal körlük, hemiparez Çoğunda tRNALeu A3243G mutasyonu var. Daha nadir 12 kadar mutasyon var. MERRF: miyoklonus epilepsi Mitokondriyal miyopati, serebellar ataxia, nöbetler. tRNALys A8344G, T8356C, G8363A mutasyonları. Bir vakada tRNAPhe G611A mutasyonu

23
Protein kodlayıcı genlerdeki defektler
ATPaz6 geni, nt-8993 TG veya TC NARP: nöropati, ataxia, retinitis pigmentoza Genç yetişkinleri etkiler, retinitis pigmentoza, demans, ataxia, sensör nöropati (mutant yük %70) MILS: maternal kalıtılan Leigh sendromu Bazal ganglia ve beyin kökünde karakteristik simetrik lezyonlar (mutant yük %90) T G değişimi ile T C değişiminden daha kötü hastalık seyri
 MILS: maternal kalıtılan Leigh sendromu. Bazal ganglia ve beyin kökünde karakteristik simetrik lezyonlar (mutant yük %90) T G değişimi ile T C değişiminden daha kötü hastalık seyri.")
24
Protein kodlayıcı genlerdeki defektler
LHON: Leber’in herediter optik nöropatisi Gençlerde akut veya subakut görme kaybı Çok sayıda mutasyon tanımlanmış ancak sadece kompleks I’ i etkileyenler patojenik. Daha ziyade erkeklerde görülüyor.

25
nDNA hasarı Solumun zincirinin komponentlerini kodlayan nDNA genlerindeki mutasyonlar İntergenomik sinyal defektleri Mitokondriyal protein transport defektleri Mitokondriyal membranın lipit kompozisyonunun defektleri Mitokondri fizyon, füzyon ve motilitesindeki defektler

26
Solunum zinciri proteinlerini etkileyen nDNA mutasyonları
Direkt Solunum zincirinin alt birimlerini etkileyen mutasyonlar (Leigh sendromunun otozomal resesif formu) Koenzim Q10 defektli hastalarda tedavi önemli İndirekt COX, SURF1, SCO2 vb… COX defektif Leigh sendromu
 Koenzim Q10 defektli hastalarda tedavi önemli. İndirekt. COX, SURF1, SCO2 vb… COX defektif Leigh sendromu.")
27
Genomlar arası sinyal defektleri
“Otozomal resesif mtDNA çoklu delesyonları” ile ortaya çıkan PEO ve diğer semptomlar Timidin fosforilaz (TP) genindeki mutasyonlar: otozomal resesif MNGIE (mitokondriyal nörogastro/intestinal ensefalomiyopati) Twinkle (helikaz) mutasyonu: otozomal dominant PEO POLG mutasyonları: otozomal dominant veya resesif PEO, psikiyatrik semptomlar, parkinsonizm
 genindeki mutasyonlar: otozomal resesif MNGIE (mitokondriyal nörogastro/intestinal ensefalomiyopati) Twinkle (helikaz) mutasyonu: otozomal dominant PEO. POLG mutasyonları: otozomal dominant veya resesif PEO, psikiyatrik semptomlar, parkinsonizm.")
28
Mitokondriyal protein transport defektleri
Çok fazla genetik bilgi yok, yaşamla bağdaşmaz? Bilinen iki defekt: TIMM8a geni Membranlar arası transport komponenti olan sağırlık-distoni proteini (DDP1) kodlar. X’ e bağlı resesif Nörosensör işitme kaybı, distoni, kortikal körlük, psikiyatrik semptomlar Otozomal dominant HSP60 gen mutasyonu: Herediter spastik parapleji
 kodlar. X’ e bağlı resesif. Nörosensör işitme kaybı, distoni, kortikal körlük, psikiyatrik semptomlar. Otozomal dominant HSP60 gen mutasyonu: Herediter spastik parapleji.")
29
İç mitokondriyal membranın lipit komponenti
X’ e bağlı resesif Barth sendromu: mitokondriyal miyopati, büyüme geriliği, kardiyopati, lökopeni Fosfolipid asetil transferaz anologu olan tafazzin kodlayan TAZ genindeki mutasyon kardiyolipin düzeyini düşürerek hastalığa yol açar.

30
Mitokondriyal motilite ve fizyon
Dinamin-ile ilişkili guanozin trifosfataz (OPA1) genindeki mutasyon: otozomal dominant optik atrofi Hastalarda lökositlerinde kümelenmiş mitokondri içeriği
 genindeki mutasyon: otozomal dominant optik atrofi. Hastalarda lökositlerinde kümelenmiş mitokondri içeriği.")
32
TANI

34
TANI Primer veya sekonder solunum zinciri bozukluğu?
Friedreich ataxia: frataxine gen mutasyonu ile demir-sülfür metabolizmasının bozulması Sekonder bozukluklar diagnostik olmak zorunda değil ama unutulmamalı!!!! Mitokondriyal-nüklear genler? Genotip-fenotip korelasyonu? Çok hasta / az mutasyon Heteroplazmi Moleküler tekniklerin yetersizliği

35
MELAS LHON Alzheimer IQ
Neredeyiz??? 1. Çok sayıda fenotipik özelliğin (patolojik veya fizyolojik) mitokondriyal kökenli olduğunu biliyoruz MELAS LHON Alzheimer IQ 2. Bu fenotipik özelliklerde etkili çok sayıda gen ve genetik düzenlenme biliyoruz
 mitokondriyal kökenli olduğunu biliyoruz. MELAS LHON Alzheimer IQ. 2. Bu fenotipik özelliklerde etkili çok sayıda gen ve genetik düzenlenme biliyoruz.")
36
Neredeyiz??? Yeni mutasyonlar (www.mitomap.org)
Homoplazmik mutasyonlar Mitokondriyal kalıtım (Schwartz M, Vissing J. Paternal inheritence of mitochondrial DNA, New Engl. J. Med. 347, , 2002.) mtDNA haplotiplerinin önemi Patojenik mekanizma
 mtDNA haplotiplerinin önemi. Patojenik mekanizma.")
37
KANSER

Benzer bir sunumlar













.>")





