Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
ÇANKIRI KARATEKİN ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
T.C. ÇANKIRI KARATEKİN ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ TRİAZOL VE SCHİFF BAZI İÇEREN TEK KRİSTAL YAPILARIN DENEYSEL VE TEORİK YÖNTEMLERLE AYDINLATILMASI GAMZE TUNAR YÜKSEK LİSANS TEZİ FİZİK ANABİLİM DAL DANIŞMAN DOÇ. DR. ÇİĞDEM YÜKSEKTEPE ATAOL

2
2. HESAPLAMALI KİMYA Hesaplamalı kimya, moleküler modelleme olarak da adlandırılabilir. Bu model, bilgisayar programı yardımıyla kimyasal problemleri incelemek için geliştirilmiştir. Teorik kimya, kimyayı matematiksel yöntemlerle tanımlar. Kimyasal yapıları ve tepkimeleri fizik kanunlarına dayanarak açıklamaya çalışır. Hesaplamalı kimya ise geliştirilen teorik yöntemleri uygular ve elde edilen sonuçları yorumlar. Böylece deneysel ve teorik sonuçlar arasında bir köprü kurar. Hesaplamalı kimya ile gözlem yolu ile elde edilmesi zor moleküller ve tepkimeler hakkında bilgi sahibi olunur. Yarıiletkenler, süper iletkenler, plastikler ve seramikler hesaplamalı kimya yardımıyla incelenmiştir [1]. Deneysel çalışmaları desteklemek ya da deneysel çalışma yapmadan teorik olarak elde edilecek sonuçları önceden tahmin edebilmek amacıyla hesaplamalı yöntemleri kullanacak araştırmacılar için dört farklı yöntem bulunmaktadır [2].

3
2.1. Moleküler Mekanik (MM) yöntemleri içeren AMPER, CHARM ve HYPERCHEM gibi programları sayabiliriz. Bu programlar bir kimyasal sistemdeki atomlar arasındaki etkileşmeleri klasik mekanik kuralları ile tanımlar. Başka bir ifade ile molekülün toplam potansiyel enerjisini minimum yapan molekül yapısı, atomlar arası etkileşmeler klasik mekanik yasalara göre hesaplanarak bulunur. Dolayısıyla hamiltoniyen operatörü ve dalga fonksiyonu kullanılmaz, hesaplamalarda elektronlar dikkate alınmaz. Bu yöntemde, etrafında küresel dağıldığı kabul edilen elektronlarla sarılmış atom çekirdekleri katı kürelere ve bu katı küreler arasındaki Coulomb etkileşmeleri de harmonik kuvvete benzetilerek, birbiriyle etkileşen küreler topluluğu dikkate alınır. Atomlar arası kimyasal bağlar ise yaylara benzetilir. Bu yöntem diğerlerine göre oldukça hızlıdır ve temel haldeki bir sistemin enerjisini tam olarak hesaplayabilir. Bir tepkime sistemi modellenerek bağ oluşumu ya da parçalanmasını içeren işlemler yapılamaz.

4
Yarı Deneysel (Semi-Emprical(SE)) yöntemler ise ab initio moleküler orbital yöntemler ile moleküler mekanik yöntemler arasında yer alır ve ab initio yöntemleri gibi kuantum mekaniksel esaslara dayanır. Bu metoda başvurulmasının nedeni, ab initio yönteminin büyük moleküller için doğru sonuçlar vermemesidir. Yarı-denel yöntemler ilk olarak organik moleküllerin yapı ve reaksiyonlarında ve organik kimya çalışmalarında kullanılmıştır daha sonra bütün moleküller için geliştirilmiştir. Kullanılan hamiltoniyen gerçek hamiltoniyenden daha basittir. Schrödinger eşitliğinin yaklaşık çözümlerini elde etmek için o sisteme uygun deneysel verilere yakın sonuçlar verebilecek parametreler kullanılır [3’]. Bu yöntemler, Hartree-Fock (HF) öz uyumlu alan (Self Consistent Field (SCF)) hesaplama yönteminin esasına dayanırlar [4’]. Yöntemlerin güvenirliği her şeyden önce parametrelerin doğru olmasına bağlıdır. Yarı-denel bazı yöntemler şunlardır: CNDO, INDO, MINDO, ZINDO, MNDO, AM1 (Austin Model), PM3 (Parametric Method). Bu yöntemleri yapısında bulunduran paket programlarından bazıları MOPAC, AMPAC, HYPERCHEM v.s. dir [3].
![Yarı Deneysel (Semi-Emprical(SE)) yöntemler ise ab initio moleküler orbital yöntemler ile moleküler mekanik yöntemler arasında yer alır ve ab initio yöntemleri gibi kuantum mekaniksel esaslara dayanır. Bu metoda başvurulmasının nedeni, ab initio yönteminin büyük moleküller için doğru sonuçlar vermemesidir. Yarı-denel yöntemler ilk olarak organik moleküllerin yapı ve reaksiyonlarında ve organik kimya çalışmalarında kullanılmıştır daha sonra bütün moleküller için geliştirilmiştir. Kullanılan hamiltoniyen gerçek hamiltoniyenden daha basittir. Schrödinger eşitliğinin yaklaşık çözümlerini elde etmek için o sisteme uygun deneysel verilere yakın sonuçlar verebilecek parametreler kullanılır [3’]. Bu yöntemler, Hartree-Fock (HF) öz uyumlu alan (Self Consistent Field (SCF)) hesaplama yönteminin esasına dayanırlar [4’]. Yöntemlerin güvenirliği her şeyden önce parametrelerin doğru olmasına bağlıdır.](http://slideplayer.biz.tr/slide/9114754/27/images/4/Yar%C4%B1+Deneysel+%28Semi-Emprical%28SE%29%29+y%C3%B6ntemler+ise+ab+initio+molek%C3%BCler+orbital+y%C3%B6ntemler+ile+molek%C3%BCler+mekanik+y%C3%B6ntemler+aras%C4%B1nda+yer+al%C4%B1r+ve+ab+initio+y%C3%B6ntemleri+gibi+kuantum+mekaniksel+esaslara+dayan%C4%B1r.+Bu+metoda+ba%C5%9Fvurulmas%C4%B1n%C4%B1n+nedeni%2C+ab+initio+y%C3%B6nteminin+b%C3%BCy%C3%BCk+molek%C3%BCller+i%C3%A7in+do%C4%9Fru+sonu%C3%A7lar+vermemesidir.+Yar%C4%B1-denel+y%C3%B6ntemler+ilk+olarak+organik+molek%C3%BCllerin+yap%C4%B1+ve+reaksiyonlar%C4%B1nda+ve+organik+kimya+%C3%A7al%C4%B1%C5%9Fmalar%C4%B1nda+kullan%C4%B1lm%C4%B1%C5%9Ft%C4%B1r+daha+sonra+b%C3%BCt%C3%BCn+molek%C3%BCller+i%C3%A7in+geli%C5%9Ftirilmi%C5%9Ftir.+Kullan%C4%B1lan+hamiltoniyen+ger%C3%A7ek+hamiltoniyenden+daha+basittir.+Schr%C3%B6dinger+e%C5%9Fitli%C4%9Finin+yakla%C5%9F%C4%B1k+%C3%A7%C3%B6z%C3%BCmlerini+elde+etmek+i%C3%A7in+o+sisteme+uygun+deneysel+verilere+yak%C4%B1n+sonu%C3%A7lar+verebilecek+parametreler+kullan%C4%B1l%C4%B1r+%5B3%E2%80%99%5D.+Bu+y%C3%B6ntemler%2C+Hartree-Fock+%28HF%29+%C3%B6z+uyumlu+alan+%28Self+Consistent+Field+%28SCF%29%29+hesaplama+y%C3%B6nteminin+esas%C4%B1na+dayan%C4%B1rlar+%5B4%E2%80%99%5D.+Y%C3%B6ntemlerin+g%C3%BCvenirli%C4%9Fi+her+%C5%9Feyden+%C3%B6nce+parametrelerin+do%C4%9Fru+olmas%C4%B1na+ba%C4%9Fl%C4%B1d%C4%B1r..jpg "Yarı-denel bazı yöntemler şunlardır: CNDO, INDO, MINDO, ZINDO, MNDO, AM1 (Austin Model), PM3 (Parametric Method). Bu yöntemleri yapısında bulunduran paket programlarından bazıları MOPAC, AMPAC, HYPERCHEM v.s. dir [3].")
5
Ab-initio Yöntemleri ise Ab-initio terimi Latincede ‘‘başlangıçtan itibaren’’ anlamına gelir. Ab-initio hesaplamalar, deneysel verileri içermeyen doğrudan teorik prensiplerden ortaya çıkan hesaplamalar için kullanılan kuantum mekaniksel yaklaşımlardır. Bu yaklaşımlar kullanılarak molekül yapısı ve buna bağlı olarak değişim gösteren parametreler hakkında önemli bilgiler elde edilir. Ab-initio hesaplamalarda en sık kullanılan yaklaşımlar, Hartree-Fock Öz Uyumlu Alan (HF-SCF), MP2 v.s. (Young, 2001) [4]. Ab-initio hesaplamalar ile çözümleme yapılırken sistemin potansiyel ve kinetik enerjilerini içeren bir Hamiltonyen işlemcisi yazılır. Schrödinger denkleminin çözümü için elde edilen Hamiltonyen işlemcisi sistemi tanımlamak için bütün verileri içerisinde barındıran bir dalga fonksiyonuna uygulanır. Ab-initio yöntemlerinde dalga fonksiyonu olarak Slater ve Gaussian tipi orbitaller kullanılır. Daha sonra varyasyon yöntemi kullanılarak sistemin enerjisinin minimum olduğu durum elde edilir. Bu sayede üzerinde hiçbir deneysel çalışma yapmadan bir molekülün geometrik yapısı ve titreşim frekansları gibi parametreler hakkında bilgi elde edilebilir. Ab-initio yöntemleri ile geniş kapsamlı teorik hesaplamalar yapmak için GAUSSIAN, GAMESS, HYPERCHEM, HONDO ve CACHE gibi bilgisayar programlar geliştirilmiştir.
![Ab-initio Yöntemleri ise Ab-initio terimi Latincede ‘‘başlangıçtan itibaren’’ anlamına gelir. Ab-initio hesaplamalar, deneysel verileri içermeyen doğrudan teorik prensiplerden ortaya çıkan hesaplamalar için kullanılan kuantum mekaniksel yaklaşımlardır. Bu yaklaşımlar kullanılarak molekül yapısı ve buna bağlı olarak değişim gösteren parametreler hakkında önemli bilgiler elde edilir. Ab-initio hesaplamalarda en sık kullanılan yaklaşımlar, Hartree-Fock Öz Uyumlu Alan (HF-SCF), MP2 v.s. (Young, 2001) [4].](http://slideplayer.biz.tr/slide/9114754/27/images/5/Ab-initio+Y%C3%B6ntemleri+ise+Ab-initio+terimi+Latincede+%E2%80%98%E2%80%98ba%C5%9Flang%C4%B1%C3%A7tan+itibaren%E2%80%99%E2%80%99+anlam%C4%B1na+gelir.+Ab-initio+hesaplamalar%2C+deneysel+verileri+i%C3%A7ermeyen+do%C4%9Frudan+teorik+prensiplerden+ortaya+%C3%A7%C4%B1kan+hesaplamalar+i%C3%A7in+kullan%C4%B1lan+kuantum+mekaniksel+yakla%C5%9F%C4%B1mlard%C4%B1r.+Bu+yakla%C5%9F%C4%B1mlar+kullan%C4%B1larak+molek%C3%BCl+yap%C4%B1s%C4%B1+ve+buna+ba%C4%9Fl%C4%B1+olarak+de%C4%9Fi%C5%9Fim+g%C3%B6steren+parametreler+hakk%C4%B1nda+%C3%B6nemli+bilgiler+elde+edilir.+Ab-initio+hesaplamalarda+en+s%C4%B1k+kullan%C4%B1lan+yakla%C5%9F%C4%B1mlar%2C+Hartree-Fock+%C3%96z+Uyumlu+Alan+%28HF-SCF%29%2C+MP2+v.s.+%28Young%2C+2001%29+%5B4%5D..jpg "Ab-initio hesaplamalar ile çözümleme yapılırken sistemin potansiyel ve kinetik enerjilerini içeren bir Hamiltonyen işlemcisi yazılır. Schrödinger denkleminin çözümü için elde edilen Hamiltonyen işlemcisi sistemi tanımlamak için bütün verileri içerisinde barındıran bir dalga fonksiyonuna uygulanır. Ab-initio yöntemlerinde dalga fonksiyonu olarak Slater ve Gaussian tipi orbitaller kullanılır. Daha sonra varyasyon yöntemi kullanılarak sistemin enerjisinin minimum olduğu durum elde edilir. Bu sayede üzerinde hiçbir deneysel çalışma yapmadan bir molekülün geometrik yapısı ve titreşim frekansları gibi parametreler hakkında bilgi elde edilebilir. Ab-initio yöntemleri ile geniş kapsamlı teorik hesaplamalar yapmak için GAUSSIAN, GAMESS, HYPERCHEM, HONDO ve CACHE gibi bilgisayar programlar geliştirilmiştir.")
6
Yoğunluk Fonksiyonel Teorisi (YFT) (Density Functional Theory (DFT)), diğer SE ve ab initio yöntemleri gibi kuantum mekaniksel esaslara dayanır fakat YFT, molekülün dalga fonksiyonunu değil elektron yoğunluk fonksiyonunu baz alarak türetilen bir hesaplamalı metotdur. ve hesaplamalı kimya araştırmacıları için hemen hemen en kullanışlı metotdur. YFT metotları metaller, yarıiletkenler ve yalıtkanların taban durum özelliklerini tanımlamak için en uygun metotdur. Bu teori, 1964 yılında Hohenberg ve Kohn tarafından ortaya atılan iki teorem ile oluşmuştur ve Kohn-Sham yaklaşıklıklarını temel alır. YFT metodunun en önemli avantajı, ab initio yöntemine kıyasla hesaplama zamanında artış olmaksızın hesaplama doğruluğunda artışın olmasıdır. YFT metotları, GAUSSIAN, GAMESS, HYPERCHEM, SPORTAN gibi pek çok popüler paket programlarda yer almaktadır [5].
 (Density Functional Theory (DFT)), diğer SE ve ab initio yöntemleri gibi kuantum mekaniksel esaslara dayanır fakat YFT, molekülün dalga fonksiyonunu değil elektron yoğunluk fonksiyonunu baz alarak türetilen bir hesaplamalı metotdur. ve hesaplamalı kimya araştırmacıları için hemen hemen en kullanışlı metotdur. YFT metotları metaller, yarıiletkenler ve yalıtkanların taban durum özelliklerini tanımlamak için en uygun metotdur. Bu teori, 1964 yılında Hohenberg ve Kohn tarafından ortaya atılan iki teorem ile oluşmuştur ve Kohn-Sham yaklaşıklıklarını temel alır. YFT metodunun en önemli avantajı, ab initio yöntemine kıyasla hesaplama zamanında artış olmaksızın hesaplama doğruluğunda artışın olmasıdır. YFT metotları, GAUSSIAN, GAMESS, HYPERCHEM, SPORTAN gibi pek çok popüler paket programlarda yer almaktadır [5]..")
7
3. MATERYAL VE YÖNTEM Moleküllerin yapılarını incelemek atomların yapılarını incelemekten çok daha zor ve karmaşıktır. Atomlarda valans olarak adlandırılan elektronların, ortak bağlar yaparak atomları birbirine bağlamaları sonucu moleküller oluşur. Çekirdeklerin kütlesi, elektronların kütlesinden çok daha büyük olduğundan elektronlara göre daha kararlı oldukları söylenebilir. Moleküllerin yapılarını çözümlemek için kuantum mekaniksel yaklaşımlara başvurulur. Kuantum mekaniksel olarak ifade edilen bir dalga fonksiyonu, fiziksel bir sistemi tanımlamak için gereken bütün verileri içermektedir. Bir atom ya da molekül için sistemin dalga fonksiyonu elde edilip, Schrödinger denklemi kesin olarak çözüldüğünde sistemin izinli enerji durumlarının tümü elde edilebilir. Sistemin toplam enerjisinin bulunabilmesi için çekirdeklerin uzaydaki konumlarının bilinmesi gerekir. Çekirdeklerin bulunmuş oldukları konumlardan yararlanarak sistemin toplam enerjisi, konumlara bağlı bir fonksiyon olarak yazılır. Sistemdeki çekirdeklerin bulunabilecekleri tüm konumlar göz önüne alınarak, sistemin toplam enerjisinin en düşük olduğu durum elde edilir. Molekülün en kararlı olduğu bu enerji değeri elde edildiğinde, molekülün bağ uzunluğu ve bağ açıları gibi geometrik parametreleri tanımlanabilir.

8
Çekirdeklerin bulunmuş oldukları konumlara göre enerjinin birinci türevi alındığında, çekirdekler üzerine etkiyen kuvvetler elde edilir. Bu kuvvetler sistemdeki atomların zamanla ne kadar yer değiştirdiğinin anlaşılmasını sağlar. Bu sayede sistemdeki atomların bağlar aracılığıyla yapmış oldukları titreşim hareketleri için titreşim frekansları hesaplanabilir (Ertaş, 2006). Çözümleme yapılması gereken sistemdeki atom sayısı arttığında, Schrödinger denklemini çözmek neredeyse imkânsız hale gelmektedir. Bu tür çok atomlu sistemleri çözümlemek için yaklaşım metotları geliştirilmiştir. Bunlar; Moleküler Mekanik, Yarı-Deneysel (ampirik), Ab-initio ve Yoğunluk Fonksiyonel Teorisi(Density Funtional Theory DFT) gibi yöntemlerdir. Bu seminerde hesaplamalı kimya yöntemlerinden biri olan Yoğunluk Fonksiyonel Teorisi ve teoride yapılan yaklaşıklıklar ayrıntılı olarak anlatılarak, YFT’den elde ettiğimiz teorik sonuçlar ile X-ışını kırınımı yönteminden elde ettiğimiz deneysel sonuçların karşılaştırılması yapılacaktır.

9
3.1 Etkileşme Halindeki Elektronlar ve Çekirdek Sistemi İçin Temel Eşitlikler
Etkileşen çekirdek ve elektronlardan oluşan bir sistemin hamiltoniyeni genel olarak şöyle ifade edilebilir: (3.1) Burada, elektronlar küçük harfli indislerle, MI kütlesine ve ZI atom numarasına sahip olan çekirdeklerde büyük harfli indislerle gösterilir. Birinci ve dördüncü terimler; sırasıyla elektron ve çekirdeğin kinetik enerjileri, ikinci terim; çekirdek ve elektronlar arasındaki Coulomb çekim etkileşimi, üçüncü terim, elektronlar arasındaki ve beşinci terim de çekirdekler arasındaki Coulomb itme etkileşimidir. Yukarıda tanımlanan sistem çok parçacık problemidir ve zamandan bağımsız Schrödinger denkleminin çözümüyle açıklanabilir: (3.2)
 Burada, elektronlar küçük harfli indislerle, MI kütlesine ve ZI atom numarasına sahip olan çekirdeklerde büyük harfli indislerle gösterilir. Birinci ve dördüncü terimler; sırasıyla elektron ve çekirdeğin kinetik enerjileri, ikinci terim; çekirdek ve elektronlar arasındaki Coulomb çekim etkileşimi, üçüncü terim, elektronlar arasındaki ve beşinci terim de çekirdekler arasındaki Coulomb itme etkileşimidir. Yukarıda tanımlanan sistem çok parçacık problemidir ve zamandan bağımsız Schrödinger denkleminin çözümüyle açıklanabilir: (3.2)")
10
Burada, çok parçacıklı sistemin dalga fonksiyonu ve E, sistemin enerjisidir. Bu problemin çözümü oldukça zordur. Bu yüzden problemi çözebilmek için bazı yaklaşımlara ihtiyaç duyulur. Bu yaklaşımlardan bir tanesi Born-Oppenheimer yaklaşımıdır. Bu yaklaşım elektron ve çekirdeklerin hareketlerinin ayrı ayrı incelenmesi ilkesine dayanır [6]. Born-Oppenheimer Yaklaşımı Bu yaklaşım, 1927 yılında yeni keşfedilmeye başlanan kuantum kimyasının ilk yıllarında Born ve Oppenheimer tarafından önerilmiş olup, hala vazgeçilmez bir şekilde kuantum kimyasında kullanılmaktadır [7]. Born-Oppenheimer yaklaşımı temelde çekirdeğin kütlesinin elektronun kütlesinden çok daha ağır olduğunu ve bu yüzden çekirdeğin hareketinin, elektronun hareketinden çok daha yavaş olduğunu ifade etmektedir.
![Burada, çok parçacıklı sistemin dalga fonksiyonu ve E, sistemin enerjisidir. Bu problemin çözümü oldukça zordur. Bu yüzden problemi çözebilmek için bazı yaklaşımlara ihtiyaç duyulur. Bu yaklaşımlardan bir tanesi Born-Oppenheimer yaklaşımıdır. Bu yaklaşım elektron ve çekirdeklerin hareketlerinin ayrı ayrı incelenmesi ilkesine dayanır [6].](http://slideplayer.biz.tr/slide/9114754/27/images/10/Burada%2C+%C3%A7ok+par%C3%A7ac%C4%B1kl%C4%B1+sistemin+dalga+fonksiyonu+ve+E%2C+sistemin+enerjisidir.+Bu+problemin+%C3%A7%C3%B6z%C3%BCm%C3%BC+olduk%C3%A7a+zordur.+Bu+y%C3%BCzden+problemi+%C3%A7%C3%B6zebilmek+i%C3%A7in+baz%C4%B1+yakla%C5%9F%C4%B1mlara+ihtiya%C3%A7+duyulur.+Bu+yakla%C5%9F%C4%B1mlardan+bir+tanesi+Born-Oppenheimer+yakla%C5%9F%C4%B1m%C4%B1d%C4%B1r.+Bu+yakla%C5%9F%C4%B1m+elektron+ve+%C3%A7ekirdeklerin+hareketlerinin+ayr%C4%B1+ayr%C4%B1+incelenmesi+ilkesine+dayan%C4%B1r+%5B6%5D..jpg "Born-Oppenheimer Yaklaşımı. Bu yaklaşım, 1927 yılında yeni keşfedilmeye başlanan kuantum kimyasının ilk yıllarında Born ve Oppenheimer tarafından önerilmiş olup, hala vazgeçilmez bir şekilde kuantum kimyasında kullanılmaktadır [7]. Born-Oppenheimer yaklaşımı temelde çekirdeğin kütlesinin elektronun kütlesinden çok daha ağır olduğunu ve bu yüzden çekirdeğin hareketinin, elektronun hareketinden çok daha yavaş olduğunu ifade etmektedir.")
11
Bu yaklaşım iki basamaktan oluşur:
Birinci basamakta, 3.1 eşitliğinde 1/MI terimi çok küçük olacağından nükleer kinetik enerjiyi temsil eden ve Tn operatörü olarak gösterilen dördüncü terim ihmal edilerek sistemin toplam hamiltoniyeninden çıkarılır. Hamiltoniyende kalan ve He olarak gösterilen elektronik hamiltoniyende, çekirdeğin konumları sabittir. Bu arada elektron-çekirdek etkileşimleri kaldırılmaz ve elektronlar, boşlukta kesin bir yerde sabitlenmiş çekirdeğin Coulomb potansiyelini hisseder. Born-Oppenheimer yaklaşımının bu birinci basamağı bu yüzden sık sık sabitlenmiş çekirdek yaklaşımı olarak da adlandırılır. Elektronik enerji özdeğeri Ee, çekirdeğin seçilen konumları olan ye bağlıdır. Bu konumları küçük basamaklarda değiştirerek ve elektronik Schrödinger denklemini çözerek, elektronik özdeğeri ‘nin bir fonksiyonu olarak bulunur. Hartree atomik birimlerini olarak kabul edersek sistemin hamilyoniyeni ,

12
‘nin bileşenlerinin kısmi türevlerini
(3.3) şeklini alır. Burada elektronik hamiltoniyeni de içine alan Schrödinger denkleminin çözümü, (3.4) şeklinde olup, 3.4 eşitliğindeki elektronik dalga fonksiyonudur. Born-Oppenheimer yaklaşımında ikinci basamak, ‘nin bileşenlerinin kısmi türevlerini içeren nükleer kinetik enerjinin tekrar getirilmesidir. Böylece nükleer hareket için, (3.5)
 şeklini alır. Burada elektronik hamiltoniyeni de içine alan Schrödinger denkleminin çözümü, (3.4) şeklinde olup, 3.4 eşitliğindeki. elektronik dalga fonksiyonudur. Born-Oppenheimer yaklaşımında ikinci basamak, ‘nin bileşenlerinin kısmi türevlerini. içeren nükleer kinetik enerjinin tekrar getirilmesidir. Böylece nükleer hareket için, (3.5)")
13
şeklinde olan Schrödinger denklemi çözülür. Çekirdeğin hamiltoniyeni;
(3.6) şeklindedir. Toplam enerji, çekirdekler arası itmeyi de içine alan, (3.7) şeklinde bir ifadeye sahiptir. Born-Oppenheimer yaklaşımı günümüzde halen yaygın bir şekilde kullanılmasına rağmen, her zaman geçerli olmayabilir. Bu yaklaşım elektron ile çekirdeğin hareketi birbirinden ayrılmadığında geçersizdir. Örneğin uyarılmış moleküllerde çekirdek o kadar hızlı hareket eder ki, elektron bu hareketi aynı anda fark edemez [8].
 şeklindedir. Toplam enerji, çekirdekler arası itmeyi de içine alan, (3.7) şeklinde bir ifadeye sahiptir. Born-Oppenheimer yaklaşımı günümüzde halen yaygın bir şekilde kullanılmasına rağmen, her zaman geçerli olmayabilir. Bu yaklaşım elektron ile çekirdeğin hareketi birbirinden ayrılmadığında geçersizdir. Örneğin uyarılmış moleküllerde çekirdek o kadar hızlı hareket eder ki, elektron bu hareketi aynı anda fark edemez [8].")
14
3.5 eşitliğinde tanımlanan elektronik yapı çözümü için birçok farklı yaklaşım kullanılır. Örneğin kuantum kimyasında kullanılan çok yaygın yaklaşımlardan bir tanesi Hartree-Fock (HF) yaklaşımıdır. Bu yaklaşımın avantajı; tek elektron dalga fonksiyonunu içeren bir slater determinantı kullanması, varyasyonel olması ve toplam enerjiyi minimize eden bir deneme dalga fonksiyonu kullanmasıdır. Fakat Hartree-Fock metodu elektronlar arasındaki korelasyonu göz önüne almaz. Yoğunluk Fonksiyoneli Teorisi (YFT) Yoğunluk fonksiyoneli teorisi (YFT)’nin temeli, 1927 yıllarda Thomas ve Fermi [9-10] tarafından yapılan çalıŞmaları temel alan Hohenberg ve Kohn teoremleri [11] ve onun devamı olan Kohn–Sham teoremlerine [12] dayanmaktadır [13]. Teorik çalışmalarda önemli bir yere sahip olan moleküler yapının belirlenmesi, enerjinin gradyenti hesaplamalarına dayanan işlemler sayesinde incelenmektedir.
 Yoğunluk fonksiyoneli teorisi (YFT)’nin temeli, 1927 yıllarda Thomas ve Fermi [9-10] tarafından yapılan çalıŞmaları temel alan Hohenberg ve Kohn teoremleri [11] ve onun devamı olan Kohn–Sham teoremlerine [12] dayanmaktadır [13]. Teorik çalışmalarda önemli bir yere sahip olan moleküler yapının belirlenmesi, enerjinin gradyenti hesaplamalarına dayanan işlemler sayesinde incelenmektedir.")
15
Birinci ve ikinci dereceden analitik gradyentlerin uygulanması karmaşık moleküllerin tam olarak optimize edilmesini mümkün kılar. Hatree-Fock metodu bu tür işlemleri Yoğunluk Fonksiyon teorisinden çok daha önce gerçekleştirmiştir. Ancak HF hesaplamaları organik moleküllerde bağ uzunlukları için tatmin edici sonuçlar verirken, organometalik bileşiklerde doğrulardan çok daha uzak sonuçlar verdiği fark edilmiştir (Ziegler, 1991). Yoğunluk fonksiyon teorisi Kuantum fiziğinin gelecek vadeden en etkili metotlarından birisidir. Bir sistemin elektronik yapısını, elektron dalga fonksiyonu yerine elektron yoğunluğuna bağlı temel bir fonksiyon olarak açık bir şekilde ifade etmeye çalışan bir teoridir.
. Yoğunluk fonksiyon teorisi Kuantum fiziğinin gelecek vadeden en etkili metotlarından birisidir. Bir sistemin elektronik yapısını, elektron dalga fonksiyonu yerine elektron yoğunluğuna bağlı temel bir fonksiyon olarak açık bir şekilde ifade etmeye çalışan bir teoridir..")
16
DFT başlangıçta sadece bir molekülün taban-durum enerjisini bulmak için kullanılıyordu. Bu teorinin pratik uygulamaları Hatree-Fock metoduna benzer yapıda bir metot ifade eden Kohn ve Sham tarafından geliştirildi. DFT metodunda elektron yoğunluğu, HF orbitallerinin matematiksel formuna benzer temel fonksiyonların bir lineer kombinasyonu olarak ifade edilir. Kohn ve Sham, HF eşitliklerindeki değiş-tokuş potansiyel teriminden farklı, sadece elektron yoğunluğunun bir fonksiyonu olan daha genel bir değiş-tokuş korelasyon (ilgisi) potansiyel terimi geliştirerek Schrödinger denklemini çözmeyi başardılar. Elektron yoğunluğunun kullanımındaki avantaj, Coulomb etkileşimi için çözülmesi gereken integrallerin sadece elektron yoğunluğu üzerinde çözülmesi ve hesaplamaların bazı elektron korelasyonlarını içermesidir. Bu sayede DFT hesaplamalarının, HF hesaplamalarından daha hızlı ve daha doğru sonuçlar verdiği söylenebilir (Kohn ve Sham, 1965; Chermette, 1998; Young, 2001).
.")
17
3.1.3. Thomas-Fermi-Dirac Yaklaşımı
Kuantum sistemlerinin ilk Yoğunluk Fonksiyonel Teorisi, Thomas ve Fermi tarafından 1927 yılında önerilen metottur [14]. Orijinal Thomas-Fermi elektron sisteminin kinetik enerjisi yoğunluğun açık bir fonksiyonelidir. Etkileşen elektron sisteminin yoğunluğu, verilen herhangi bir noktadaki yerel yoğunluğa eşit olan etkileşmeyen elektronların oluşturduğu bir homojen gaz şeklinde ideal hale getirmiştir. Thomas ve Fermi elektronlar arasındaki değiş-tokuş ve korelasyonu ihmal etmiştir. Dirac ise bugün halen kullanımda olan değiş-tokuş için yerel yaklaşımı geliştirerek formüle etmiştir [15, 16].

18
Thomas-Fermi yaklaşımı elementlerin durum denklemine uygulanmıştır
Thomas-Fermi yaklaşımı elementlerin durum denklemine uygulanmıştır. Fakat Thomas-Fermi tipi yaklaşımlar moleküllerin bağlanması ve atomların kabuk yapıları gibi bazı fiziksel ve kimyasal gerçeklerden yoksundur. Bu nedenle madde içerisinde elektronların anlamlı bir tanımlamasında başarısız olur. Hohenberg-Kohn Teoremleri 1964 yılında Hohenberg ve Kohn, çok parçacık sistemini tam olarak çözen Yoğunluk Fonksiyonel Teorisini formüle etmişlerdir. Bu formülasyon bir dış potansiyeli altında sabitlenmiş çekirdek alanında hareket eden elektronların etkileştiği herhangi bir sisteme uygulanabilir. Bu durumda sistemin hamiltoniyeni, (3.11) şeklinde olur. Yoğunluk fonksiyonel teorisi, Hohenberg ve Kohn tarafından iki teorem üzerine oturtulmuştur.
 şeklinde olur. Yoğunluk fonksiyonel teorisi, Hohenberg ve Kohn tarafından iki teorem üzerine oturtulmuştur.")
19
1. Teorem : Bir dış potansiyeli altında etkileşen parçacıkların sistemi için,
potansiyeli, taban durum yoğunluğu tarafından tam olarak tanımlanır. 1. Sonuç : Bu yüzden hamiltoniyen tamamiyle belirlendiği için taban ve uyarılmış haldeki tüm durumlar için çok parçacık dalga fonksiyonları belirlenir. Bu nedenle sistemin tüm özellikleri, sadece verilen taban durum yoğunluğu ile tümüyle belirlenir. 2. Teorem : Herhangi bir dış potansiyel için geçerli olan seçilen yoğunluğuna bağlı, enerji için bir E[n] fonksiyoneli tanımlanabilir. Herhangi bir özel bir için sistemin tam taban durumu enerjisi, bu fonksiyonelin minimum değeridir ve fonksiyoneli minimize eden , sistemin taban durum parçacık yoğunluğu ’ dir. 2. Sonuç : E[n] fonksiyoneli tam taban durumu enerjisi ve yoğunluğunu belirlemek için yeterlidir. Genellikle elektronların uyarılmış durumları diğer araçlarla belirlenir.

20
Kohn-Sham Yaklaşımı Kohn-Sham yaklaşımında amaç, 3.1 eşitliğindeki hamiltoniyeni sağlayan etkileşen çok parçacık sistemini daha farklı ve daha kolay çözülebilen bir sistemle yer değiştirmektir [17]. Kohn-Sham yaklaşımı, gerçek etkileşen sistemin taban durum yoğunluğunu, seçilen etkileşmeyen bir sistemin taban durum yoğunluğuna eşit kabul eder. Bu etkileşmeyen bir sistem için bağımsız parçacık denklemlerine yol açar ve bu denklemler, yoğunluğun bir değiş-tokuş korelasyon fonksiyonelinin içine yerleştirilmiş çok parçacık terimleri ile tam olarak çözülebilir. Denklemlerin çözümü ile gerçek etkileşen sistemin taban durum yoğunluğu ve enerjisi, değiş-tokuş korelasyon fonksiyonelinde kullanılan yaklaşımlarla sınırlandırılmış bir doğruluk içerisinde belirlenebilir.

21
Kohn ve Sham büyük moleküler sistemlerin ve yoğun maddenin özelliklerinin, ilk prensiplerinden yola çıkılarak tahmin edilmesine yönelik pek çok hesaplamanın temelini oluşturan çok kullanışlı yaklaşımlar önermiştir. Kohn-Sham yaklaşımı iki kabul üzerine kuruludur: 1. Gerçek etkileşen sistemin taban durum yoğunluğu, etkileşmeyen parçacıklardan oluşan yapay sistemin taban durum yoğunluğu ile ifade edilir. 2. Yapay hamilyoniyen, genel kinetik enerji operatörüne ve r noktasında spini σ olan elektron üzerine etkiyen etkin bir yerel potansiyel ’ ye sahip olacak şekilde seçilir. Bu yerel form, Kohn-Sham yaklaşımının belirleyici karakteristiklerine sahip olan son derece kullanışlı bir basitleştirmedir. Gerçek hesaplamalar, aşağıda verilen yapay hamiltoniyen ile tanımlanan yapay bağımsız parçacık sistemi üzerine gerçekleştirilir. (3.12)
")
22
Yapay sistemin yoğunluğu her bir spin orbitallerinin karelerinin toplamı ile verilir. Yani yapay sistemin yoğunluğu, (3.13) şeklindedir. Bağımsız parçacık kinetik enerjisi Ts ise, (3.14) şeklinde verilir ve yoğunluğunun klasik Coulomb etkileşme enerjisi; (3.15) şeklinde tanımlanır. Tamamen etkileşen çok parçacık problemi için Kohn-Sham yaklaşımındaki amaç, taban durumu enerji fonksiyoneli için Hohenberg-Kohn ifadesini, (3.16)
 şeklindedir. Bağımsız parçacık kinetik enerjisi Ts ise, (3.14) şeklinde verilir ve yoğunluğunun klasik Coulomb etkileşme enerjisi; (3.15) şeklinde tanımlanır. Tamamen etkileşen çok parçacık problemi için Kohn-Sham yaklaşımındaki amaç, taban durumu enerji fonksiyoneli için Hohenberg-Kohn ifadesini, (3.16)")
23
yada daha açık bir şekilde,
şeklinde yazmaktır. Burada , çekirdek ve diğer elektronlar için bir dış potansiyeldir ve EII ise çekirdekler arası etkileşim enerjisidir. Bağımsız parçacık kinetik enerjisi Ts, belirgin bir şekilde orbitallerin bir fonksiyoneli şeklinde verilir. Bununla birlikte her bir spin σ için Ts, Hohenberg-Kohn düşüncelerinin eşitlik 3.12 ile verilen bağımsız parçacık hamiltoniyenine uygulanması ile yoğunluğunun yegane bir fonksiyoneli olmak zorundadır. Değiş-tokuş ve korelasyonun tüm çok parçacık etkileri, değiş-tokuş korelasyon enerjisi Exc içerisinde gruplandırılır. Toplam enerji için olan ifadeler Exc’ nin Hohenberg-Kohn fonksiyoneli şeklinde aşağıda verildiği gibi yazılabileceğini gösterir. (3.17) yada daha açık bir şekilde, (3.18)
 yada daha açık bir şekilde, (3.18)")
24
3.1.6. Kohn-Sham Varyasyonel Denklemleri
Taban durumu için Kohn-Sham yardımcı sisteminin çözümü, ya yoğunluk ’ ye yada etkin potansiyel ’ye göre minimizasyon problemi gibi incelenebilir eşitliğinden de görüldüğü gibi Ts açıkça orbitallerin bir fonksiyoneli gibi açıklandığı için diğer tüm terimler yoğunluğun fonksiyonelleri olarak düşünülebilir. Dalga fonksiyonları değiştirilebilir ve varyasyonel denklemi türetmek için zincir kuralı kullanılabilir. (3.19) Tabi ki burada ortonormalizasyon şartları sağlanmalıdır. (3.20)
 Tabi ki burada ortonormalizasyon şartları sağlanmalıdır. (3.20)")
25
Bu, Rayleigh-Ritz prensibine denktir [19, 20]. 3. 12 ve 3
Bu, Rayleigh-Ritz prensibine denktir [19, 20] ve 3.13 eşitliklerini, ve Ts için kullanarak, (3.21) ve Lagrange çarpan metodu kullanılarak Kohn-Sham denklemi Schrödinger benzeri denkleme dönüşür. (3.22) Burada ’ ler özdeğerlerdir ve HKS’ de Hartree atomik birimleri içinde etkin homitoniyendir.
![Bu, Rayleigh-Ritz prensibine denktir [19, 20] ve 3](http://slideplayer.biz.tr/slide/9114754/27/images/25/Bu%2C+Rayleigh-Ritz+prensibine+denktir+%5B19%2C+20%5D+ve+3.jpg "Bu, Rayleigh-Ritz prensibine denktir [19, 20] ve 3.13 eşitliklerini, ve Ts için kullanarak, (3.21) ve Lagrange çarpan metodu kullanılarak Kohn-Sham denklemi Schrödinger benzeri denkleme dönüşür. (3.22) Burada ’ ler özdeğerlerdir ve HKS’ de Hartree atomik birimleri içinde etkin homitoniyendir.")
26
(3.23) ve (3.24) şeklindedir. 3.22 ve 3.23eşitlikleri Kohn-Sham denklemleri olarak bilinir. Denklemler, sonuçlanan yoğunluk ile kendi içinde tutarlı olması gereken bir potansiyel ile bağımsız parçacık denklemlerinin şekline sahiptir. Bu denklemler, herhangi bir yaklaşımdan bağımsız olan fonksiyonel Exc[n] biliniyorsa, etkileşen sistem için tam taban durum yoğunluğunun ve enerjisinin bulunmasını sağlar. Böylece Hohenberg-Kohn teoremlerinden bulunan durum yoğunluğu, minimumdaki potansiyeli tam olarak belirler ve verilen herhangi bir etkileşen elektron sistemi ile ilişkilendirilmiş tek bir Kohn-Sham potansiyeli ortaya çıkar.

27
ise; şeklinde tanımlanır.
Değiş-Tokuş Korelasyon Potansiyeli (Vxc) Değiş-tokuş korelasyon potansiyeli değiş-tokuş korelasyon enerjisi Exc’ nin fonksiyonel türevidir. Değiş-tokuş korelasyon enerjisi Exc ; (3.25) şeklinde tanımlanır ve buradaki noktasının bazı komşulukları içinde sadece yoğunluk ’ ya bağlı olan, noktasında elektron başına düşen enerjidir. Değiş-tokuş korelasyon potansiyeli ise; (3.26) şeklinde tanımlanır.
 Değiş-tokuş korelasyon potansiyeli değiş-tokuş korelasyon enerjisi Exc’ nin fonksiyonel türevidir. Değiş-tokuş korelasyon enerjisi Exc ; (3.25) şeklinde tanımlanır ve buradaki noktasının bazı komşulukları içinde sadece yoğunluk ’ ya bağlı olan, noktasında elektron başına düşen enerjidir. Değiş-tokuş korelasyon potansiyeli. ise; (3.26) şeklinde tanımlanır.")
28
Yoğunluk Fonksiyonel Teorisi, pratik ve yaklaşık fonksiyonellerin başarısından dolayı günümüzde çokça kullanılan bir metottur. Yoğunluğun bir fonksiyoneli olarak tanımlanan değiş-tokuş korelasyon enerjisi Exc[n], Kohn-Sham yaklaşımı içerisinde çok önemli bir niceliktir. Kohn-Sham yaklaşımının iki temel üstünlüğü vardır: Birincisi, yaklaşım, etkileşen çok parçacık problemini çözmeyi, bağımsız parçacık denklemlerini çözmekten daha kolay kılar. İkincisi ve belki de daha önemlisi, bağımsız parçacık kinetik enerjisi ve uzun sıralı Hartree terimlerini açıkça ayırarak, kalan değiş-tokuş korelasyon fonksiyoneli Exc[n]’ in makul bir şekilde yoğunluğun yerel veya hemen hemen yerel bir fonksiyonele yaklaştırılabilmesidir. Kesin fonksiyonel Exc[n] oldukça karmaşık olmasına rağmen, esas işlem kayda değer basit yaklaşımlar ile yapılır. Bu yaklaşımlar Yerel Yoğunluk (YYY), Genelleştirilmiş Gradyent Yaklaşımı(GGY) ve Hibrit Fonksiyonellerdir.
![Yoğunluk Fonksiyonel Teorisi, pratik ve yaklaşık fonksiyonellerin başarısından dolayı günümüzde çokça kullanılan bir metottur. Yoğunluğun bir fonksiyoneli olarak tanımlanan değiş-tokuş korelasyon enerjisi Exc[n], Kohn-Sham yaklaşımı içerisinde çok önemli bir niceliktir.](http://slideplayer.biz.tr/slide/9114754/27/images/28/Yo%C4%9Funluk+Fonksiyonel+Teorisi%2C+pratik+ve+yakla%C5%9F%C4%B1k+fonksiyonellerin+ba%C5%9Far%C4%B1s%C4%B1ndan+dolay%C4%B1+g%C3%BCn%C3%BCm%C3%BCzde+%C3%A7ok%C3%A7a+kullan%C4%B1lan+bir+metottur.+Yo%C4%9Funlu%C4%9Fun+bir+fonksiyoneli+olarak+tan%C4%B1mlanan+de%C4%9Fi%C5%9F-toku%C5%9F+korelasyon+enerjisi+Exc%5Bn%5D%2C+Kohn-Sham+yakla%C5%9F%C4%B1m%C4%B1+i%C3%A7erisinde+%C3%A7ok+%C3%B6nemli+bir+niceliktir..jpg "Kohn-Sham yaklaşımının iki temel üstünlüğü vardır: Birincisi, yaklaşım, etkileşen çok parçacık problemini çözmeyi, bağımsız parçacık denklemlerini çözmekten daha kolay kılar. İkincisi ve belki de daha önemlisi, bağımsız parçacık kinetik enerjisi ve uzun sıralı Hartree terimlerini açıkça ayırarak, kalan değiş-tokuş korelasyon fonksiyoneli Exc[n]’ in makul bir şekilde yoğunluğun yerel veya hemen hemen yerel bir fonksiyonele yaklaştırılabilmesidir. Kesin fonksiyonel Exc[n] oldukça karmaşık olmasına rağmen, esas işlem kayda değer basit yaklaşımlar ile yapılır. Bu yaklaşımlar Yerel Yoğunluk (YYY), Genelleştirilmiş Gradyent Yaklaşımı(GGY) ve Hibrit Fonksiyonellerdir.")
29
3.1.8. Hibrit Fonksiyonelleri
Hibrit fonksiyonelleri, YFT de değiş-tokuş ve korelasyon enerji fonksiyonelini bulmak için kullanılan fonksiyonellerdir. Hibrit fonksiyoneller birçok fonksiyonellerin bir karışımıdır. Burada yer alan Hartree-Fock değiş- tokuş enerjisi gerçek Hartree-Fock enerjisi değildir. Aşağıdaki ifade de yer alan orbitaller, Kohn-Sham orbitalleri olduğu için bu değiş-tokuş enerjisi HF-tipi değiş-tokuş enerjisidir. Şöyle ki; (3.27) Pek çok hibrit fonksiyonelleri tanımlanmıştır. Bunlardan en popüler olan hibrit fonksiyoneli, 1993 yılında Becke (B3) tarafından keşfedilen değiş-tokuş enerji fonksiyoneli ve 1988 yılında Lee-Yang-Parr (LYP) tarafından keşfedilen korelasyon enerji fonksiyonelidir ve bu değiş-tokuş korelasyon fonksiyoneli Becke3LYP veya B3LYP fonksiyonelidir ve 3.28 eşitliği ile ifade edilebilir: (3.28)
 Pek çok hibrit fonksiyonelleri tanımlanmıştır. Bunlardan en popüler olan hibrit fonksiyoneli, 1993 yılında Becke (B3) tarafından keşfedilen değiş-tokuş enerji fonksiyoneli ve 1988 yılında Lee-Yang-Parr (LYP) tarafından keşfedilen korelasyon enerji fonksiyonelidir ve bu değiş-tokuş korelasyon fonksiyoneli Becke3LYP veya B3LYP fonksiyonelidir ve 3.28 eşitliği ile ifade edilebilir: (3.28)")
30
Burada yer alan ilk terim, gradiyent düzeltmesi içermeyen değiş-tokuş fonksiyoneli, ikinci terim, KS orbitallerini temel alan HF-tipi değiş-tokuş fonksiyoneli, son üç terim ise, sırasıyla, Becke88 değiş-tokuş, Vosko-Wilk-Nusair korelasyon ve Lee-Yang-Parr korelasyon fonksiyonelleridir. Enerjiler önündeki parametreler ise moleküler atomizasyon enerjisi için hesaplanan en uyumlu katsayılardır. Başka bir hibrit fonksiyonel ise Perdew, Burke ve Ernzerhof (PBE) tarafından ifade edilen PBE1PBE fonksiyonelidir eşitliği ile ifade edilebilir: (3.29) Buradaki ilk terim, Perdew, Burke ve Ernzerhof (PBE) değiş-tokuş, ikinci terim HF-tipi değiş-tokuş ve son terim ise PBE korelasyon fonksiyonelleridir. Gerek YY gerekse GGY gerekse de Hibrit fonksiyonellerin hepsinin amacı değiş-tokuş korelasyon enerjisini dolayısıyla da potansiyeli hesaplamaktır. Bunlar için de bazı mümkün baz fonksiyonları tanımlanmıştır. Bunlar BLYP, PBEPBE, B3LYP, PBE1PBE v.s. şeklinde tanımlanabilir. B3LYP, organik bileşikler için iyi metaller için kötü sonuçlar verirken, BLYP, metaller için iyi organik bileşikler için kötü sonuçlar verir [24].
 Buradaki ilk terim, Perdew, Burke ve Ernzerhof (PBE) değiş-tokuş, ikinci terim HF-tipi değiş-tokuş ve son terim ise PBE korelasyon fonksiyonelleridir. Gerek YY gerekse GGY gerekse de Hibrit fonksiyonellerin hepsinin amacı değiş-tokuş korelasyon enerjisini dolayısıyla da potansiyeli hesaplamaktır. Bunlar için de bazı mümkün baz fonksiyonları tanımlanmıştır. Bunlar BLYP, PBEPBE, B3LYP, PBE1PBE v.s. şeklinde tanımlanabilir. B3LYP, organik bileşikler için iyi metaller için kötü sonuçlar verirken, BLYP, metaller için iyi organik bileşikler için kötü sonuçlar verir [24].")
31
BAZ SETLERİ Baz setleri, moleküldeki atomların değerlik orbitallerinin lineer bir kombinasyonu olup, moleküler orbitallere karşılık gelen matematiksel fonksiyonların bir setidir. Farklı veya aynı cins atomların atomik orbitallerinin lineer kombinasyonu moleküler orbitalleri verdiği bilindiği gibi N tane atomik baz fonksiyonunun lineer birleşiminden N tane moleküler orbital türetilir [25]. Çok farklı çeşitte baz setleri olup, bir hesaplama için doğru bir baz setinin seçimi hesaplamalı kimyada önemli bir karardır. Dalga fonksiyonu, bir elektronun veya elektron grubunun çekirdekle veya birbirleriyle ilişkisinin matematiksel bir tarifidir. Dalga fonksiyonun karesi ise elektronların mümkün olabileceği tüm yerler üzerinden toplamını hesaplar ve elektron veya elektronların bulunma olasılıklarını bize verir. Bulunma olasılığı, doğrudan moleküllerin enerjilerine ve diğer karakteristik özelliklerine dönüştürülebilir. Dört farklı baz seti vardır.

32
Bunlar; 1. Minimal Baz Setleri (STO-1G, STO-2G,....) 2. Split Valans Baz Setleri (3-21G, 6-31G....) 3. Polarize Baz Setleri(3-21G(d,p), 6-311G(d,p).....) 4. Diffuse Baz Setleri(3-21++G,.....) Bir baz seti seçimine hesaplama tipine bakılarak karar verilir. Gerçekte mükemmel bir baz seti, Schrödinger eşitliğinin tam bir çözümünü ortaya koyacaktır. Sonuç olarak; Yoğunluk Fonksiyonel Teoride kullanılan bazı baz setlerini tablo halinde verebiliriz [26].
, 6-311G(d,p).....) 4. Diffuse Baz Setleri(3-21++G,.....) Bir baz seti seçimine hesaplama tipine bakılarak karar verilir. Gerçekte mükemmel bir baz seti, Schrödinger eşitliğinin tam bir çözümünü ortaya koyacaktır. Sonuç olarak; Yoğunluk Fonksiyonel Teoride kullanılan bazı baz setlerini tablo halinde verebiliriz [26].")
33
Tablo 3.1 Baz Setlerinin Serisi
Baz Setlerinin Adı Tanımı STO-3G Minimal baz setidir, büyük sistemler üzerinde daha çok nitel sonuçlar için kullanılır. 3-21G 6-31G İki kat zeta (split valans baz seti); valans bölgedeki fonksiyonların 2 seti orbitallerin daha doğru tarifini gösterir. 6-31G* yada 6-31G (d) Polarize split valans baz setidir. Ağır atomlara polarizasyon fonksiyonunu ekler; en çok orta büyüklükteki sistemler için kullanılır. 6-31G** yada 6-31G (d, p) İki kez polarize split valans baz setidir. Hidrojenlere de polarizasyon fonksiyonunu ekler; enerji hesaplamalarında kesin doğru sonuçlar için kullanılır. 6-31+G* yada 6-31+G (d) Diffuse polarize split valans baz setidir; uyarılmış durum, anyonlar için önemlidir. 6-31+G** yada 6-31+G (d, p) Diffuse iki kez polarize split valans baz setidir. 6-311+G** yada 6-311+G (d, p) Üç kat zeta; baz setine ekstra valans elektronlarını ekler. Sonuç olarak, ilgili molekülün enerjisi ve geometrik parametreleri YFT modelinde öz uyumlu alan yöntemine ile hesaplanır ve sıralama aşağıdaki gibidir.
; valans bölgedeki fonksiyonların 2 seti orbitallerin daha doğru tarifini gösterir. 6-31G* yada. 6-31G (d) Polarize split valans baz setidir. Ağır atomlara polarizasyon fonksiyonunu ekler; en çok orta büyüklükteki sistemler için kullanılır. 6-31G** yada. 6-31G (d, p) İki kez polarize split valans baz setidir. Hidrojenlere de polarizasyon fonksiyonunu ekler; enerji hesaplamalarında kesin doğru sonuçlar için kullanılır G* yada G (d) Diffuse polarize split valans baz setidir; uyarılmış durum, anyonlar için önemlidir G** yada G (d, p) Diffuse iki kez polarize split valans baz setidir G** yada G (d, p) Üç kat zeta; baz setine ekstra valans elektronlarını ekler. Sonuç olarak, ilgili molekülün enerjisi ve geometrik parametreleri YFT modelinde öz uyumlu alan yöntemine ile hesaplanır ve sıralama aşağıdaki gibidir.")
34
. Şekil 3.2 Bir kristalin toplam enerjisini kendini doğrulama metodunu kullanarak hesaplayan bir bilgisayar programının akış diyagramı

35
. Hesaplanan bu cvi lerden tekrar moleküler orbitaller elde edilir.
. Yaklaşık bir moleküler orbital ifadesi, giriş değeri olarak tahmin edilir. Bu tahmin atomik orbitallerin çizgisel kombinasyonuna dayanır. Atomik orbitaller olarak baz setleri seçilir. . Elektron yoğunluğu bu tahmini moleküler orbitalden hesaplanır ve giriş değeri olarak kabul edilir. . Tahmini enerji ifadesi hesaplanır. . HKS hamiltoniyeni hesaplanır. . Kohn-Sham denklemlerindeki enerji özdeğeri ve cvi katsayıları hesaplanır. . Hesaplanan bu cvi lerden tekrar moleküler orbitaller elde edilir. Bu başlangıç değer hesaplamalarından sonra öz uyumlu alan çevrimi tekrar başlar. Yani elektron yoğunluğu ve yukarıda ifade edilen bütün terimler hesaplanır. Bu işlem hesaplanan bu büyüklüklerin bir önceki değeri ile hesaplanan değeri arasındaki fark kabul edilir bir seviyeye inene kadar devam ettirilir.

36
3.1.10. Geometrik Optimizasyon
Bu bölümde moleküllerde denge durum geometrisinin nasıl hesaplandığı üzerinde durulacaktır. İncelenecek yöntem gradyent optimizasyonu veya kuvvet metodu olarak bilinmektedir. Hesaplamalar, moleküler sistem belirli bir geometride iken yapılır. Moleküllerdeki yapısal değişiklikler molekülün enerjisinde ve diğer birçok özelliğinde değişiklikler oluşturur. Molekülün yapısındaki küçük değişiklikler sonucu enerjinin koordinata bağımlılığı "potansiyel enerji yüzeyi (PES)" olarak tanımlanır. Potansiyel enerji yüzeyi moleküler yapı ile sonuç enerji arasındaki ilişkidir. Bir molekül için potansiyel enerji eğrileri veya yüzeyi bilinirse denge durumundaki geometriye karşılık gelen minimum enerjili nokta bulunabilir. İki atomlu bir molekülde bağ gerilmesine karşılık gelen elektronik enerji grafiği sekil 3.3.deki gibi verilebilir. Şekilde minimum enerjili nokta Em ve Xm ile gösterilmektedir. Potansiyelin harmonik kısmı Hooke yasası ile verilir.
 olarak tanımlanır. Potansiyel enerji yüzeyi moleküler yapı ile sonuç enerji arasındaki ilişkidir. Bir molekül için potansiyel enerji eğrileri veya yüzeyi bilinirse denge durumundaki geometriye karşılık gelen minimum enerjili nokta bulunabilir. İki atomlu bir molekülde bağ gerilmesine karşılık gelen elektronik enerji grafiği sekil 3.3.deki gibi verilebilir. Şekilde minimum enerjili nokta Em ve Xm ile gösterilmektedir. Potansiyelin harmonik kısmı Hooke yasası ile verilir.")
37
göre ikinci türevidir ve kuvvet sabiti olarak adlandırılır.
(3.30) Burada G enerjinin konuma göre ikinci türevidir ve kuvvet sabiti olarak adlandırılır. Yani kuvvet sabiti, (3.31) ifadesi ile verilir. Sekil 3.3. iki atomlu bir molekülde elektronik enerjinin atomlar arası mesafeye bağlılığı (Csizmadia, 1990) [27].
 Burada G enerjinin konuma. göre ikinci türevidir ve kuvvet sabiti olarak adlandırılır. Yani kuvvet sabiti, (3.31) ifadesi ile verilir. Sekil 3.3. iki atomlu bir molekülde elektronik enerjinin atomlar arası mesafeye bağlılığı (Csizmadia, 1990) [27].")
38
Çok boyutlu problemlerde genelleştirilmiş Hooke yasası,
(3.32) olarak ifade edilir. Burada yer değiştirme vektörü ve G elemanlarını köşegen ve köşegen dışı etkilesen kuvvet sabitlerinin oluşturduğu Hessian matrisi adını alır. (3.33)
 olarak ifade edilir. Burada yer değiştirme vektörü ve G elemanlarını köşegen ve köşegen dışı etkilesen kuvvet sabitlerinin oluşturduğu Hessian matrisi adını alır. (3.33)")
39
Moleküler geometri optimizasyonu
konumlarına karşılık gelen minimum enerjili noktaları bulmak demektir. Bu ilk aşamada gradyent vektörü g’ yi bulmak demektir. (3.34) İkinci aşamada ise gradyent vektörünün sıfır olduğu noktaları bulmaktır. (3.35)
 İkinci aşamada ise gradyent vektörünün sıfır olduğu noktaları bulmaktır. (3.35)")
40
Sekil 3.4. İki boyutta potansiyel enerji yüzeyi
Daha önce belirtildiği gibi gradyent vektörünün sıfır olduğu noktalar minimum enerjili duruma karşılık gelir ve molekülün bu durumdaki geometrisi denge durumu geometrisidir. Bir molekül için potansiyel enerji yüzeyinde bir çok maksimum ve minimumlar görülür. (Sekil 3.4.). Potansiyel enerji yüzeyindeki minimumlar sistemin dengede olduğu yerlerdir. Tek bir molekül için farklı minimumlar farklı konformasyonlara veya yapısal izomerlere karşılık gelir. Sırtlardaki düşük nokta bir yönde yerel minimum, diğer yönde bir maksimumdur. Bu tür noktalara "eyer noktaları, saddle point" adı verilir. Eyer noktaları iki denge yapısı arasındaki geçiş yapısına karşılık gelir. Sekil İki boyutta potansiyel enerji yüzeyi
. Potansiyel enerji yüzeyindeki minimumlar sistemin dengede olduğu yerlerdir. Tek bir molekül için farklı minimumlar farklı konformasyonlara veya yapısal izomerlere karşılık gelir. Sırtlardaki düşük nokta bir yönde yerel minimum, diğer yönde bir maksimumdur. Bu tür noktalara eyer noktaları, saddle point adı verilir. Eyer noktaları iki denge yapısı arasındaki geçiş yapısına karşılık gelir. Sekil 3.4. İki boyutta potansiyel enerji yüzeyi.")
41
Geometri optimizasyonları genellikle potansiyel enerji yüzeyindeki minimumları araştırır, bunun neticesinde de moleküler sistemlerin denge yapılarını tahmin eder. Optimizasyon aynı zamanda geçiş yapılarını da araştırır. Minimumlarda ve eyer noktalarında enerjinin birinci türevi yani gradyent sıfırdır. Kuvvet de gradyentin negatifidir; bu nedenle bu noktalarda kuvvet de sıfırdır. Potansiyel enerji yüzeyinde gradyent vektörü g’ nin sıfır olduğu noktaya "kararlı noktalar" adı verilir. Tüm başarılı geometri optimizasyonları bu kararlı noktaları bulmayı hedefler. Geometri optimizasyonu giriş (başlangıç) geometrisindeki moleküler yapıdan başlayarak potansiyel enerji yüzeyini dolaşır. Bu noktada enerji ve gradyenti hesaplar ve hangi yöne doğru ne kadar gidileceğine karar verir. Gradyent eğimin dikliğini verdiği kadar, yüzey boyunca mevcut noktadan enerjinin çok hızlı düstügü noktayı da verir. Enerjinin atomik koordinatlara göre ikinci türevi kuvvet sabitini verir. Optimizasyon algoritmalarının çoğu kuvvet sabitleri matrisi olarak bilinen Hessianı da hesaplar veya tahmin eder. Kuvvet sabitleri bu noktadaki yüzeyin eğriliğini tanımlar ki bu bir sonraki aşamanın belirlenmesinde ek bilgi verir. Optimizasyon yakınsadığında tamamlanmış olur. Yani hesaplanan geometride g vektörü sıfır ve bir sonraki aşamada hesaplanan geometrik parametrelerin değerleri ile hesaplanan değerler arasındaki fark ihmal edilebilir bir değerde ise optimizasyon tamamlanmış olur (Dadakdeniz, 2007; Doğru, 2007). [28].
. [28].")
42
4.1. Triazol ve Schiff Bazı İçeren Tek Kristal Yapıların Deneysel ve Teorik Yöntemlerle Aydınlatılması X- ışını kırınım yöntemi ile moleküler yapısı belirlenen bileşiklerin kuantum mekanik ve moleküler mekanik metotlarla optimize edilip; elektronik ve yapısal parametrelerin deneysel verilerle karşılaştırılması, son yıllarda fizikçi ve kimyacıların çalıştıkları başlıca konular arasında yer almaktadır. Kristal yapı analizinde deneysel yöntemlerden biri olan X-ışını kırınım verileri kullanılarak moleküllerin geometrik yapıları, bağ uzunlukları, bağ açıları ve torsiyon açıları yapılabilmektedir. Ayrıca kristal yapıların istiflenme şekilleri de paket programlarla analiz edilebilmektedir. Bulunan sonuçlar teorik hesaplamalar ile karşılaştırılarak desteklenmektedir.

43
Biz C22H24N4OS ve C27H37N3 kristal yapıların analizini, tek kristal difraktometresinden elde edilen şiddet verilerini WinGX paket programını kullanarak yapmaya çalıştık. Teorik hesaplamalarımızda ise Gaussian03W programını kullandık. Teorik hesaplamalarımızda DFT/B3LYP 6-311G(d,p) baz setini kullanarak yapıların geometrik optimizasyonunu yaptırdık. Bu çalışmada, deneysel ve teorik olarak elde edilen bağ uzunlukları, bağ açıları ve torsiyon açıları karşılaştırılmış ve ayrıca hidrojen bağ yetenekleri incelenmiş, geometrik ve fiziksel özelliklerinin literatürde daha önce sentezlenen benzer bileşiklerle karşılaştırılması yapılmıştır. Aşağıda yapıları aydınlatılan kristallerin içerdiği gruplar hakkında kısaca bilgiler yer almaktadır.

50
(4-Etil-5-piridin-4-yl-4H-[1,2,4]triazol-3-ylsulfanil)-1-(3-metil-3-fenil-siklobütil)-etanbir Tek Kristalinin Analizi Yapısında Triazol bulunduran C22H24N4OS tek kristalinin şematik görünümü Triazollerin Genel Özellikleri Triazol halka sistemi 1885 Yılında Bladin tarafından bulunmuştur. Beşli halkada üç azot atomu içeren bileşiklere ‘Triazoller’ adı verilir. Beşli halkada üç azot atomu içeren bu bileşikler, halkadaki azot atomlarının birbirine karşı durumuna göre 1,2,3-triazol (vic-triazol) (1) ve 1,2,4-triazol (sim-triazol) (2) olmak üzere birbirinin izomeri olan iki triazol halkası olarak adlandırılabilir. Triazoller aromatik bileşiklerdir.
![(4-Etil-5-piridin-4-yl-4H-[1,2,4]triazol-3-ylsulfanil)-1-(3-metil-3-fenil-siklobütil)-etanbir Tek Kristalinin Analizi](http://slideplayer.biz.tr/slide/9114754/27/images/50/%284-Etil-5-piridin-4-yl-4H-%5B1%2C2%2C4%5Dtriazol-3-ylsulfanil%29-1-%283-metil-3-fenil-siklob%C3%BCtil%29-etanbir+Tek+Kristalinin+Analizi.jpg "Yapısında Triazol bulunduran C22H24N4OS tek kristalinin şematik görünümü Triazollerin Genel Özellikleri. Triazol halka sistemi 1885 Yılında Bladin tarafından bulunmuştur. Beşli halkada üç azot atomu içeren bileşiklere ‘Triazoller’ adı verilir. Beşli halkada üç azot atomu içeren bu bileşikler, halkadaki azot atomlarının birbirine karşı durumuna göre 1,2,3-triazol (vic-triazol) (1) ve 1,2,4-triazol (sim-triazol) (2) olmak üzere birbirinin izomeri olan iki triazol halkası olarak adlandırılabilir. Triazoller aromatik bileşiklerdir.")
51
Şekil 4.1 1,2,3-Triazol ve 1,2,4-Triazol
1,2,3- Triazol ,2,4- Triazol Şekil ,2,3-Triazol ve 1,2,4-Triazol Literatürde triazol türevleriyle yapılan çalışmalarda antimikrobiyal[29], virostatik[30], sitostatik[31], antienflamatuvar[32], analjezik[33], antikalzunvan[34], merkezi sinir sistemi depresanı[35], antihistaminik[36], hipotansif[37], diüretik[38], herbisit[39], antihelmintik[40], antifungal[41], pestisit[42], ve insektisit[43] etkili bileşiklere ulaşılmıştır. Bohm ve Karow[44], farmakolajik etki verebilen triazol türevleri üzerinde yapılan araştırmaları bir “Derleme” de toplamıştır.

52
Triazol çekirdeği içeren herhangi bir doğal bileşiğe rastlanamamıştır
Triazol çekirdeği içeren herhangi bir doğal bileşiğe rastlanamamıştır. Ancak triazol yapısı, pek çok bileşiğin yapısında yer alan ve bazı önemli fizyolojik olaylarda rol oynayan maddelerin (Histamin, Histedin, B12 vitamini) yapısında bulunur. C22H24N4OS ve C27H38N3 tek kristal yapıları Fırat Üniversitesinde Kimya Laboratuarında sentezlenmiş olup kırınım verileri ise Ondokuz Mayıs ve Giresun Üniversitesinden elde edilmiştir.
 yapısında bulunur. C22H24N4OS ve C27H38N3 tek kristal yapıları Fırat Üniversitesinde Kimya Laboratuarında sentezlenmiş olup kırınım verileri ise Ondokuz Mayıs ve Giresun Üniversitesinden elde edilmiştir.")
53
Şekil 4.3 C22H24N4OS molekülünün Ortep şekli.
Aşağıda, X- ışını kırınımı yönteminden elde edilen C22H24N4OS tek kristalinin ORTEP görünümü verilmiştir. Şekil 4.3 C22H24N4OS molekülünün Ortep şekli. Şekil’den de göründüğü gibi kristal yapı benzen halkası, siklobütan halkası, triazol ve piridin halkasını içermektedir. Tablo 4.4.’ den de görüldüğü gibi tek kristal yapı monoklinik kristal sisteminde C2/c uzay grubuna sahiptir.

54
Tablo 4.4. C22H24N4OS tek kristalinin kristalografik datası
Kimyasal Formülü Moleküler Ağırlığı Sıcaklık, T (K) Dalga boyu (Å) Kristal Sistem Kristal Boyutu (mm3) Uzay Grubu a (Å) b (Å) c (Å) α (°) β (°) γ (°) Hacim, V (Å3) Z Tmin , Tmaks Hesaplanan Yoğunluk (Mg m-3) θ aralığı (º) Data Aralığı Ölçülen Yansımalar Bağımsız Yansımalar Gözlenen Yansımalar ( I>2σ ) Yerleştirme Faktörü R1 indisi ( I>2σ ) wR2 indisi ( I>2σ ) Δρmin, Δρmaks (e/Å3) C22H24N4OS 392.51 296 Monoklinik 0.320 x x 0.760 C 2/c (16) 5.7123(2) (15) 90 (21) 4052.7(3) 8 0.8803, 1.29 1.86 – 26.75 h= -45→45, k= -7→7, l= -38→40 12509 4293 3426 0.994 0.03 0.09 -0.216, 0.212
 Dalga boyu (Å) Kristal Sistem. Kristal Boyutu (mm3) Uzay Grubu. a (Å) b (Å) c (Å) α (°) β (°) γ (°) Hacim, V (Å3) Z. Tmin , Tmaks. Hesaplanan Yoğunluk (Mg m-3) θ aralığı (º) Data Aralığı. Ölçülen Yansımalar. Bağımsız Yansımalar. Gözlenen Yansımalar ( I>2σ ) Yerleştirme Faktörü. R1 indisi ( I>2σ ) wR2 indisi ( I>2σ ) Δρmin, Δρmaks (e/Å3) C22H24N4OS Monoklinik x x C 2/c (16) (2) (15) (21) (3) , – h= -45→45, k= -7→7, l= -38→ ,")
55
Piridin A(C1-C2-C3-N1-C4-C5), triazol B(N1-C6-N3-N4-C7) ve fenil D(C17-C22) halkaları düzlemsel yapıdadır. Siklobütan halkası C(C12-C15) düzlemsel yapıda değildir. Bu düzlemler arasındaki dihedral açılar sırasıyla şöyledir; A/B= 37.24(1)°, B/C=62.13(1)° ve C/D=38.03(1)°. Siklobütan halkasındaki C13/C14/C15 düzlemi ile C13/C12/C15 düzlemi arasındaki dihedral açı ise 25.55(3)° dir. Bulunan dihedral açı değeri literatürdeki 23.5◦ , 18.92(15)◦ , ve 19.26(17)◦ değerlerden biraz da olsa büyük olduğu gözlenmiştir. Bunun sebebi ise yapıdaki sterik etkileşmelerdir. DFT B3LYP/6-311G(d,p) baz seti seçilerek yapılan geometrik optimizasyon sonucunda elde edilen şekil ise aşağıdaki gibidir; Şekil 4.5 C22H24N4OS molekülünün Gaussview görünümü
 baz seti seçilerek yapılan geometrik optimizasyon sonucunda elde edilen şekil ise aşağıdaki gibidir; Şekil 4.5 C22H24N4OS molekülünün Gaussview görünümü.")
56
X- ışını kırınım yönteminden elde ettiğimiz sonuçlar ile yoğunluk fonksiyonel teorisinden elde ettiğimiz bağ uzunlukları, bağ açıları ve torsiyon açılarının karşılaştırması Tablo 4.5’ de verilmiştir. Tablo’dan da görüldüğü gibi B3LYP/6–311G(d, p) baz seti seçilerek yapılan hesaplama sonucunda yapının bağ uzunlukları ve bağ açıları deneysel sonuçlarla oldukça uyumlu fakat torsiyon açılarında ise az da olsa fark olduğu bulunmuştur. Bu farkın sebebi ise deneysel ölçümlerin katı fazda teorik hesaplamaların ise gaz fazında yapılmasından kaynaklandığı söylenebilir.
 baz seti seçilerek yapılan hesaplama sonucunda yapının bağ uzunlukları ve bağ açıları deneysel sonuçlarla oldukça uyumlu fakat torsiyon açılarında ise az da olsa fark olduğu bulunmuştur. Bu farkın sebebi ise deneysel ölçümlerin katı fazda teorik hesaplamaların ise gaz fazında yapılmasından kaynaklandığı söylenebilir..")
57
Tablo 4.6 C22H24N4OS molekülünün bağ uzunluğu, bağ açıları ve torsiyon açıları
Deneysel B3LYP 6–311G(d, p) Bağ Uzunlukları (Å) O1–C11 C11–C10 S1–C10 S1–C7 N2–C7 N4–C7 N4–N3 N3–C6 N2–C6 N2–C8 C8–C9 N1–C4 N1–C3 Bağ Açıları (°) C12–C11–O1 C12–C11–C10 O1–C11–C10 C11–C10–S1 S1–C7–N4 S1–C7–N2 C7–N2–C6 C7–N2–C8 N2–C8–C9 1.206(17) 1.510(2) 1.806(16) 1.744(15) 1.369 (16) 1.311(18) 1.374(18) 1.316(17) 1.361(17) 1.476(16) 1.506(2) 1.332(2) 1.328(2) 122.76(14) 114.99(12) 122.24(14) 115.98(10) 123.92(11) 125.64(10) 104.44(11) 125.75(12) 112.59(12) 1.209 1.529 1.829 1.766 1.374 1.313 1.364 1.320 1.378 1.471 1.527 1.339 1.334 122.63 114.52 122.84 115.7 124.9 124.25 103.77 125.65 113.38 devamı Deneysel B3LYP 6–311G(d, p) C7–N4–N3 N4–N3–C6 N3–C6–N2 N3–C6–C1 C6–N2–C8 C3–N1–C4 Torsiyon Açıları (°) C15–C12–C11−C10 C13–C12–C11−C10 C15–C12–C11−O1 C13–C12–C11−O1 C12–C11–C10−S1 O1–C11–C10–S1 C11–C10–S1−C7 C10–S1–C7–N4 C10–S1–C7–N2 S1–C7–N4–N3 S1–C7–N2–C6 S1–C7–N2–C8 C7–N2–C8–C9 N4–N3–C6–C1 107.31(11) 107.34(11) 110.45(12) 121.74(13) 129.70(12) 116.28(15) -77.25(16) 179.21(13) 101.26(19) -2.3(2) 174.18(11) -4.3(2) 82.50(12) 70.77(13) (12) (10) 179.46(10) 2.95(19) -80.20(17) 179.73(12) 107.31 108.13 109.91 122.50 129.78 116.69 -84.47 172.46 94.72 -8.35 170.07 -9.11 73.47 59.62 179.17 8.65 -84.40
 Bağ Uzunlukları (Å) O1–C11. C11–C10. S1–C10. S1–C7. N2–C7. N4–C7. N4–N3. N3–C6. N2–C6. N2–C8. C8–C9. N1–C4. N1–C3. Bağ Açıları (°) C12–C11–O1. C12–C11–C10. O1–C11–C10. C11–C10–S1. S1–C7–N4. S1–C7–N2. C7–N2–C6. C7–N2–C8. N2–C8–C (17) 1.510(2) 1.806(16) 1.744(15) (16) 1.311(18) 1.374(18) 1.316(17) 1.361(17) 1.476(16) 1.506(2) 1.332(2) 1.328(2) (14) (12) (14) (10) (11) (10) (11) (12) (12) devamı. Deneysel. B3LYP. 6–311G(d, p) C7–N4–N3. N4–N3–C6. N3–C6–N2. N3–C6–C1. C6–N2–C8. C3–N1–C4. Torsiyon Açıları (°) C15–C12–C11−C10. C13–C12–C11−C10. C15–C12–C11−O1. C13–C12–C11−O1. C12–C11–C10−S1. O1–C11–C10–S1. C11–C10–S1−C7. C10–S1–C7–N4. C10–S1–C7–N2. S1–C7–N4–N3. S1–C7–N2–C6. S1–C7–N2–C8. C7–N2–C8–C9. N4–N3–C6–C (11) (11) (12) (13) (12) (15) (16) (13) (19) -2.3(2) (11) -4.3(2) 82.50(12) 70.77(13) (12) (10) (10) 2.95(19) (17) (12)")
58
Simetri kodu: (i):1/2 -x, -1/2+y, 1/2-z
C22H24N4OS tek kristali, bir molekül içi bir de moleküller arası olmak üzere iki hidrojen bağına sahiptir. Molekül içi hidrojen bağı Şekil 4.4’ de görülmektedir. Tablo 6.3’ de bu hidrojen bağlarının türleri ve bağ mesafeleri verilmiştir. Şekil 4.7‘ de ise moleküller arası C−H…N türü hidrojen bağının kristalin istiflenmesinde nasıl etki yaptığı görülmektedir. (x, y, z) simetri koduna sahip siklobütan halkasının donör atomuna (C12) bağlı H12 atomu (1/2 -x, -1/2+y, 1/2-z) simetri koduna sahip başka bir molekülün triazol halkasındaki N4 atomu ile moleküller arası hidrojen bağı yapmaktadır ve bu bağ kristal boyunca [010] ve [0-10] doğrultularında devam ederek kristal istiflenmeyi oluşturmaktadır. Tablo 4.7 C22H24N4OS Tek Kristalinin Hidrojen Bağ Etkileşimleri (Å, º) Hidrojen Bağları (Å,°) D–H H...A D...A D–H...A C12–H12...N4i C8−H8B...O1 0.98 0.97 2.47 2.44 3.418(2) 3.310(4) 162 149 Simetri kodu: (i):1/2 -x, -1/2+y, 1/2-z
 simetri koduna sahip siklobütan halkasının donör atomuna (C12) bağlı H12 atomu (1/2 -x, -1/2+y, 1/2-z) simetri koduna sahip başka bir molekülün triazol halkasındaki N4 atomu ile moleküller arası hidrojen bağı yapmaktadır ve bu bağ kristal boyunca [010] ve [0-10] doğrultularında devam ederek kristal istiflenmeyi oluşturmaktadır. Tablo 4.7 C22H24N4OS Tek Kristalinin Hidrojen Bağ Etkileşimleri (Å, º) Hidrojen Bağları (Å,°) D–H. H...A. D...A. D–H...A. C12–H12...N4i. C8−H8B...O (2) 3.310(4) Simetri kodu: (i):1/2 -x, -1/2+y, 1/2-z")
59
Şekil 4.8 C22H24N4OS tek kristalinin birim hücredeki hidrojen bağı

60
(E)-1-(2-(3-mesitil-3-metilsiklobütil)-2-(2-fenilhidrazon)etil)piperadin Tek Kristalinin Analizi Yapısında Schiff bazı bulunduran C27H37N3 tek kristalinin şematik görünümü Schiff Bazlarının Genel Özellikleri İlk olarak 1869 yılında Alman Kimyacı H. Schiff tarafından sentezlenen bu bileşikler ‘Schiff Bazı’ olarak adlandırılmaktadır.

61
–C=N– grubu içeren Schiff bazları (–C=N–NH: hidrazon), zayıf bazik özellikte olup renkli kristal yapılı bileşiklerdir. Kristal yapıda olmalarından dolayı keskin erime noktasına sahiptirler. Kompleksleri ise normal şartlar altında kararlı olup çok yüksek erime noktasına sahiptirler. Schiff bazlarının kararlılığı, azometin (–C=N–) grubu ile ilgilidir. Karbon azot atomları arasındaki çift bağ ne kadar kararlı ise Schiff bazları da o kadar kararlıdır. Çünkü, –C=N– çift bağı, aromatik halka ile rezonansa girerek daha kararlı yapı meydana getirir (Turgut, 1997). Son 30 yıldır Schiff bazlarının metal komplekslerine ilgi artmış olup, fizyolojik aktivite göstermeleri, anti-tümör ve anti-mikrobik aktiviteye sahip olmaları ve eczacılıkta kullanılmaları nedeniyle oldukça önem kazanmışlardır.
 grubu ile ilgilidir. Karbon azot atomları arasındaki çift bağ ne kadar kararlı ise Schiff bazları da o kadar kararlıdır. Çünkü, –C=N– çift bağı, aromatik halka ile rezonansa girerek daha kararlı yapı meydana getirir (Turgut, 1997). Son 30 yıldır Schiff bazlarının metal komplekslerine ilgi artmış olup, fizyolojik aktivite göstermeleri, anti-tümör ve anti-mikrobik aktiviteye sahip olmaları ve eczacılıkta kullanılmaları nedeniyle oldukça önem kazanmışlardır.")
62
Koordinasyon bileşiklerinin sentezinde ligand olarak kullanılan Schiff bazları, metal atomuna birden fazla donör (verici) atomu ile bağlanarak çok dişli ligandlar sınıfına girmekte ve geçiş metalleriyle kararlı kompleksler oluşturmaktadır. Farklı donör atomu içeren Schiff bazları, metal iyonuna karşı duyarlı ve seçici olduğu için analitik uygulamalarda kullanılmaktadır (Dubey ve Yard, 1991) [45]. Bununla birlikte Schiff bazları, madeni yağlarda bulunan ve istenmeyen az miktardaki elementlerin giderilmesinde, boya, kauçuk, ilaç sanayisinde ve ziraai amaçlarla da kullanılmaktadır. Ayrıca boyar madde ve polimer teknolojisinde, ilaç sanayinde, tıpta, tarım alanında, roket yakıtı hazırlanmasında, biyolojik olayların açıklanmasında ve daha birçok alanda bu bileşiklerden büyük ölçüde yararlanılmakta ve yeni sentezlerin yapılması yönündeki çalışmalar yoğun bir şekilde devam etmektedir.
 [45]. Bununla birlikte Schiff bazları, madeni yağlarda bulunan ve istenmeyen az miktardaki elementlerin giderilmesinde, boya, kauçuk, ilaç sanayisinde ve ziraai amaçlarla da kullanılmaktadır. Ayrıca boyar madde ve polimer teknolojisinde, ilaç sanayinde, tıpta, tarım alanında, roket yakıtı hazırlanmasında, biyolojik olayların açıklanmasında ve daha birçok alanda bu bileşiklerden büyük ölçüde yararlanılmakta ve yeni sentezlerin yapılması yönündeki çalışmalar yoğun bir şekilde devam etmektedir..")
63
Şekil 4.10 C27H37N3 molekülünün Ortep şekli.
Aşağıda, X- ışını kırınımı yönteminden elde edilen C27H37N3 tek kristalinin ORTEP görünümü verilmiştir. Şekil 4.10 C27H37N3 molekülünün Ortep şekli. Şekil’den de göründüğü gibi kristal yapı benzen halkaları, siklobütan halkası, hidrazon (Schiff Bazı) ve piperadin halkasını içermektedir. Tablo 4.10.’ dan da görüldüğü gibi tek kristal yapı monoklinik kristal sisteminde P 21/n uzay grubuna sahiptir.
 ve piperadin halkasını içermektedir. Tablo 4.10.’ dan da görüldüğü gibi tek kristal yapı monoklinik kristal sisteminde P 21/n uzay grubuna sahiptir.")
64
Tablo 1 (1)’ in kristalografik datası
Kimyasal Formülü Moleküler Ağırlığı Sıcaklık, T (K) Dalgaboyu (Å) Kristal Sistem Kristal Boyutu (mm3) Uzay Grubu a (Å) b (Å) c (Å) α (°) β (°) γ (°) Hacim, V (Å3) Z Tmin , Tmaks Hesaplanan Yoğunluk (Mg m-3) θ aralığı (º) Data Aralığı Ölçülen Yansımalar Bağımsız Yansımalar Gözlenen Yansımalar ( I>2σ ) Yerleştirme Faktörü R1 indisi ( I>2σ ) wR2 indisi ( I>2σ ) Δρmin, Δρmaks (e/Å3) C27H37N3 404.6 296 Monoklinik 0.320 x x 0.760 P 21/n 9.2480(4) (5) (10) 90 97.180(4) (18) 4 0.8330, 1.000 1.10 3.13 – 27.79 h= -11→11, k= -8→13, l= -30→25 9517 4852 2676 1.043 0.06 0.14 -0.165, 0.234
 Dalgaboyu (Å) Kristal Sistem. Kristal Boyutu (mm3) Uzay Grubu. a (Å) b (Å) c (Å) α (°) β (°) γ (°) Hacim, V (Å3) Z. Tmin , Tmaks. Hesaplanan Yoğunluk (Mg m-3) θ aralığı (º) Data Aralığı. Ölçülen Yansımalar. Bağımsız Yansımalar. Gözlenen Yansımalar ( I>2σ ) Yerleştirme Faktörü. R1 indisi ( I>2σ ) wR2 indisi ( I>2σ ) Δρmin, Δρmaks (e/Å3) C27H37N Monoklinik x x P 21/n (4) (5) (10) (4) (18) , – h= -11→11, k= -8→13, l= -30→ ,")
65
Fenil B(C22-C27) ve diğer fenil D(C7-C12) halkaları düzlemsel yapıdadır. Piperadin A(N3-C17-C18-C19-C20-C21) ve siklobütan halkası C(C2-C5) düzlemsel yapıda değildirler. Bu düzlemler arasındaki dihedral açılar sırasıyla şöyledir; A/B= 66.45(1)°, B/C=66.21(1)° ve C/D=40.79(2)°. Siklobütan halkasındaki C3/C4/C5 düzlemi ile C3/C2/C5 düzlemi arasındaki dihedral açı ise 26.41(1)° dir. Bulunan dihedral açı değeri literatürdeki 23.5◦, 18.92(15)◦ , ve 19.26(17)◦ değerlerden biraz büyüktür. DFT B3LYP/6-311G(d,p) baz seti seçilerek yapılan geometrik optimizasyon sonucunda elde edilen şekil ise aşağıdaki gibidir;
 baz seti seçilerek yapılan geometrik optimizasyon sonucunda elde edilen şekil ise aşağıdaki gibidir;")
66
Şekil 4.11 C27H37N3 molekülünün Gaussview görünümü
X- ışını kırınım yönteminden elde ettiğimiz sonuçlar ile yoğunluk fonksiyonel teorisinden elde ettiğimiz bağ uzunlukları, bağ açıları ve torsiyon açılarının karşılaştırması Tablo 4.11’ de verilmiştir. Tablo’dan da görüldüğü gibi B3LYP/6–311G(d, p) baz seti seçilerek yapılan hesaplama sonucunda yapının bazı parametrelerinde biraz farklılık olduğu gözlenmiştir. Bu farkın sebebi ise deneysel ölçümlerin katı fazda teorik hesaplamaların ise gaz fazında yapılmasından kaynaklandığı söylenebilir.
 baz seti seçilerek yapılan hesaplama sonucunda yapının bazı parametrelerinde biraz farklılık olduğu gözlenmiştir. Bu farkın sebebi ise deneysel ölçümlerin katı fazda teorik hesaplamaların ise gaz fazında yapılmasından kaynaklandığı söylenebilir.")
67
Tablo 2 X-Işını ve DFT metodu ile elde edilen geometrik parametreler
Deneysel B3LYP 6–311G(d, p) Bağ Uzunlukları (Å) C2–C1 C1–N1 N1–N2 N2–C22 C1–C16 C16–N3 N3–C17 N3–C21 Bağ Açıları (°) C5–C2–C1 C3–C2–C1 C2–C1–N1 C2–C1–C16 C1–N1–N2 N1–N2–C22 N2–C22–C23 1.485(3) 1.281(3) 1.380(3) 1.382(3) 1.511(3) 1.466(3) 1.458(3) 1.462(3) 120.2(2) 117.5(2) 118.3(2) 116.2(2) 117.0(2) 118.8(2) 118.2(2) 1.504 1.285 1.355 1.393 1.522 1.470 1.469 120.1 119.3 118.5 116.7 119.2 120.4 devamı Deneysel B3LYP 6–311G(d, p) Bağ Uzunlukları (Å) N2–C22–C27 N1–C1–C16 C1–C16–N3 C16–N3–C21 C16–N3–C17 Torsiyon Açıları (°) C3–C2–C1−N1 C3–C2–C1−C16 C5–C2–C1−N1 C5–C2–C1−C16 C2–C1–N1−N2 C2–C1–C16–N3 C1–N1–N2−C22 N1–N2–C22–C23 N1–N2–C22–C27 C1–C16–N3–C17 C1–C16–N3–C21 123.5(2) 125.3(2) 115.7(2) 110.9(2) 111.5(2) 95.5(3) -84.8(3) -9.0(3) 170.7(2) -179.1(2) 137.0(2) 168.2(2) -169.9(2) 11.7(4) -63.4(3) 172.4(2) 122.4 124.6 115.3 111.0 112.1 101.7 -82.4 -4.4 171.4 178.4 135.8 176.6 -171.8 9.6 -64.6 170.0
 Bağ Uzunlukları (Å) C2–C1. C1–N1. N1–N2. N2–C22. C1–C16. C16–N3. N3–C17. N3–C21. Bağ Açıları (°) C5–C2–C1. C3–C2–C1. C2–C1–N1. C2–C1–C16. C1–N1–N2. N1–N2–C22. N2–C22–C (3) 1.281(3) 1.380(3) 1.382(3) 1.511(3) 1.466(3) 1.458(3) 1.462(3) 120.2(2) 117.5(2) 118.3(2) 116.2(2) 117.0(2) 118.8(2) 118.2(2) devamı. Deneysel. B3LYP. 6–311G(d, p) Bağ Uzunlukları (Å) N2–C22–C27. N1–C1–C16. C1–C16–N3. C16–N3–C21. C16–N3–C17. Torsiyon Açıları (°) C3–C2–C1−N1. C3–C2–C1−C16. C5–C2–C1−N1. C5–C2–C1−C16. C2–C1–N1−N2. C2–C1–C16–N3. C1–N1–N2−C22. N1–N2–C22–C23. N1–N2–C22–C27. C1–C16–N3–C17. C1–C16–N3–C (2) 125.3(2) 115.7(2) 110.9(2) 111.5(2) 95.5(3) -84.8(3) -9.0(3) 170.7(2) (2) 137.0(2) 168.2(2) (2) 11.7(4) -63.4(3) 172.4(2)")
68
Tablo 3 (1) Bileşiğinin Hidrojen Bağ Etkileşimleri (Å, º)
C27H37N3 tek kristali, iki molekül içi hidrojen bağına sahiptir, moleküller arası hidrojen bağı olmamakla birlikte C−H…Cg (π…ring) etkileşimi vardır. Molekül içi hidrojen bağları Şekil 4.11’ de görülmektedir. Tablo 3.1’ de bu hidrojen bağlarının türleri ve bağ mesafeleri verilmiştir. Şekil 4.12‘ de ise moleküller arası π…ring etkileşimi kristalin istiflenmesinde etkilidir. (x, y, z) simetri koduna sahip siklobütan halkasının donör atomuna (C5) bağlı H5B atomu 3/2-x, 1/2+y, 1/2-z simetri koduna sahip başka bir molekülün fenil (C7-C12) halka merkezi ile hidrojen bağı yapmaktadır ve bu bağ kristal boyunca [010] ve [0-10] doğrultularında devam ederek kristal istiflenmeyi oluşturmaktadır. Tablo 3 (1) Bileşiğinin Hidrojen Bağ Etkileşimleri (Å, º) Hidrojen Bağları (Å,°) D–H H...A D...A D–H...A N2–H2...N3 C5−H5A...N1 C5−H5B...Cg3i 0.86 0.97 2.22 2.55 2.69 2.760(3) 2.875(3) 3.599(3) 121 100 156 Simetri kodu : 3/2-x, 1/2+y, 1/2-z Cg(3)= (C7-C12)
 etkileşimi vardır. Molekül içi hidrojen bağları Şekil 4.11’ de görülmektedir. Tablo 3.1’ de bu hidrojen bağlarının türleri ve bağ mesafeleri verilmiştir. Şekil 4.12‘ de ise moleküller arası π…ring etkileşimi kristalin istiflenmesinde etkilidir. (x, y, z) simetri koduna sahip siklobütan halkasının donör atomuna (C5) bağlı H5B atomu 3/2-x, 1/2+y, 1/2-z simetri koduna sahip başka bir molekülün fenil (C7-C12) halka merkezi ile hidrojen bağı yapmaktadır ve bu bağ kristal boyunca [010] ve [0-10] doğrultularında devam ederek kristal istiflenmeyi oluşturmaktadır. Tablo 3 (1) Bileşiğinin Hidrojen Bağ Etkileşimleri (Å, º) Hidrojen Bağları (Å,°) D–H. H...A. D...A. D–H...A. N2–H2...N3. C5−H5A...N1. C5−H5B...Cg3i (3) 2.875(3) 3.599(3) Simetri kodu : 3/2-x, 1/2+y, 1/2-z Cg(3)= (C7-C12)")
69
Şekil 4.12 C27H37N3 tek kristalinin birim hücredeki hidrojen bağı

70
5. KAYNAKLAR [1]. Lewars E., Computational Chemistry, Kluwer Academic Publisher, 2003. [2]. Çelik I., Akkurt M., Ide S., Tutar A ve Çakmak A., G. Ü. Journal of Science 16(1):27-35, 2003. [3]. Mitchell, A.D., ''British chemical nomenclature'', E. Arnold Co., London, 1160 (1948 [4]. Young, D.C., Computational Chemistry A Practical Guide for Applying Techniques to Real-World Problems (Electronics). John Wiley and Sons, 381p, New York.). [3’]. Levine I. R., Physical Chemistry, McGraw-Hill international Editions, 1995. [4’]. Hatipoğlu A., Kuantum Mekaniksel Yapı ve Enerji Hesaplamaları, Y.T.Ü. Fen Bilimleri Enstitüsü, 2001 [5]. Atkins P. Friedman R., Molecular Quantum Mechanics, Oxford Üniversity pres., 2005. [6]. Erkişi A., Bazı Bileşiklerin Titreşim ve Elektronik Özelliklerinin Yoğunluk Fonksiyonel Teorisi ile İncelenmesi, G. Ü. Fen Bilimleri Enstitüsü, 2007. [7]. Born, M. And Oppenheimer, J. R., Zur Quantentheorie der Molekeln, Ann. Physik, 84: , 1927.
![5. KAYNAKLAR [1]. Lewars E., Computational Chemistry, Kluwer Academic Publisher,](http://slideplayer.biz.tr/slide/9114754/27/images/70/5.+KAYNAKLAR+%5B1%5D.+Lewars+E.%2C+Computational+Chemistry%2C+Kluwer+Academic+Publisher%2C.jpg "[2]. Çelik I., Akkurt M., Ide S., Tutar A ve Çakmak A., G. Ü. Journal of Science 16(1):27-35, [3]. Mitchell, A.D., British chemical nomenclature , E. Arnold Co., London, (1948. [4]. Young, D.C., Computational Chemistry A Practical Guide for Applying. Techniques to Real-World Problems (Electronics). John Wiley and Sons, 381p, New York.). [3’]. Levine I. R., Physical Chemistry, McGraw-Hill international Editions, [4’]. Hatipoğlu A., Kuantum Mekaniksel Yapı ve Enerji Hesaplamaları, Y.T.Ü. Fen Bilimleri Enstitüsü, [5]. Atkins P. Friedman R., Molecular Quantum Mechanics, Oxford Üniversity pres., [6]. Erkişi A., Bazı Bileşiklerin Titreşim ve Elektronik Özelliklerinin Yoğunluk Fonksiyonel Teorisi ile İncelenmesi, G. Ü. Fen Bilimleri Enstitüsü, [7]. Born, M. And Oppenheimer, J. R., Zur Quantentheorie der Molekeln, Ann. Physik, 84: ,")
71
[8]. Szabo, A. And Ostlund, N. S
[8]. Szabo, A. And Ostlund, N. S., Modern Quantum Chemistry, McGraw-Hill, New York, 43-44, 1989. [9]. Fermi, E., (1927). A Statistical Method for Determining some Properties of the Atom. Rend. Accad. Lincei, 6, 602. [10]. Thomas, L. H. (1927). The calculation of atomic fields. in Mathematical Proceedings of the Cambridge Philosophical Society. Cambridge Univ Press. [11]. Hohenberg, P. and W. Kohn, (1964). Inhomogeneous electron gas. Physical review, 136(3B), B864. [12]. Kohn, W. and L.J. Sham, (1965). Self-consistent equations including exchange and correlation effects. Physical Review, 140(4A), A1133. [13]. Candan, A., (2011). Kübik yapıdaki Co2MnX (X=Al, Ga, Ge, Si) Heusler alaşımlarının Yoğunluk Fonksiyonel Teorisi ile incelenmesi, Yüksek Lisans Tezi, Gazi Üniversitesi Fen Bilimleri Enstitüsü, Ankara. [14]. Lundqvist, S. And March, N. H., Theory of the Inhomogeneous Electron Gas, Plenum Pres, New York, , 1983. [15]. Lewars E., Computational Chemistry, Kluwer Academic Publisher, 2003. [16]. Dirac, P. A. M. Note on Exchange phenomena in the Thomas- Fermi atom, Proc. Cambridge Phil. Roy. Soc., 26: , 1930.
![[8]. Szabo, A. And Ostlund, N. S](http://slideplayer.biz.tr/slide/9114754/27/images/71/%5B8%5D.+Szabo%2C+A.+And+Ostlund%2C+N.+S.jpg "[8]. Szabo, A. And Ostlund, N. S., Modern Quantum Chemistry, McGraw-Hill, New York, 43-44, [9]. Fermi, E., (1927). A Statistical Method for Determining some Properties of the Atom. Rend. Accad. Lincei, 6, 602. [10]. Thomas, L. H. (1927). The calculation of atomic fields. in Mathematical Proceedings of the Cambridge Philosophical Society. Cambridge Univ Press. [11]. Hohenberg, P. and W. Kohn, (1964). Inhomogeneous electron gas. Physical review, 136(3B), B864. [12]. Kohn, W. and L.J. Sham, (1965). Self-consistent equations including exchange and correlation effects. Physical Review, 140(4A), A1133. [13]. Candan, A., (2011). Kübik yapıdaki Co2MnX (X=Al, Ga, Ge, Si) Heusler alaşımlarının Yoğunluk Fonksiyonel Teorisi ile incelenmesi, Yüksek Lisans Tezi, Gazi Üniversitesi Fen Bilimleri Enstitüsü, Ankara. [14]. Lundqvist, S. And March, N. H., Theory of the Inhomogeneous Electron Gas, Plenum Pres, New York, , [15]. Lewars E., Computational Chemistry, Kluwer Academic Publisher, [16]. Dirac, P. A. M. Note on Exchange phenomena in the Thomas- Fermi atom, Proc. Cambridge Phil. Roy. Soc., 26: ,")
72
[17]. Kohn, W. And Sham, L. J., Self-Consistent Equations Including Exchange and Correlation Efects, Phys. Rev., 140:A1133-A1138, 1965. [18]. Imada, M., Fujimori, A. And Tokura, Y., Metal-insulator transitions, Rev. Mod. Phys., 70: , 1998. [19]. Strutt, J. W., Theory of Sound, Reprint: Dover Publications, New York, 1:88, 1945. 20]. Ritz, W., Über eine neune Methode zur Losung Gewisser Varitionprobleme der mathemetischen Physik, Reine Angew. Math., 135:1-61, 1908. [21]. Perdew, J. P. And Levy, M., Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities, Phys. Rev. Lett., 51: , 1983. [22]. Sham, L. J. Nad Schlüter, M., Density-funtional theory of the energy gap, Phys. Rev. Lett., 51: , 1983. [23]. Almbladh, C. And von Barth, U., Exact results fort he charge and spin densities, exhange-correlation potentials and density-functional eigenvalues, Phys. Rev. B, 31: ,1985. [24]. Cramer, C., Essentials of Computational Chemistry, Wiley, 2007. [25]. Lewars E., Computational Chemistry, Kluwer Academic Publisher, 2003.
![[17]. Kohn, W. And Sham, L. J., Self-Consistent Equations Including Exchange and Correlation Efects, Phys. Rev., 140:A1133-A1138, 1965.](http://slideplayer.biz.tr/slide/9114754/27/images/72/%5B17%5D.+Kohn%2C+W.+And+Sham%2C+L.+J.%2C+Self-Consistent+Equations+Including+Exchange+and+Correlation+Efects%2C+Phys.+Rev.%2C+140%3AA1133-A1138%2C+1965..jpg "[18]. Imada, M., Fujimori, A. And Tokura, Y., Metal-insulator transitions, Rev. Mod. Phys., 70: , [19]. Strutt, J. W., Theory of Sound, Reprint: Dover Publications, New York, 1:88, ]. Ritz, W., Über eine neune Methode zur Losung Gewisser Varitionprobleme der mathemetischen Physik, Reine Angew. Math., 135:1-61, [21]. Perdew, J. P. And Levy, M., Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities, Phys. Rev. Lett., 51: , [22]. Sham, L. J. Nad Schlüter, M., Density-funtional theory of the energy gap, Phys. Rev. Lett., 51: , [23]. Almbladh, C. And von Barth, U., Exact results fort he charge and spin densities, exhange-correlation potentials and density-functional eigenvalues, Phys. Rev. B, 31: ,1985. [24]. Cramer, C., Essentials of Computational Chemistry, Wiley, [25]. Lewars E., Computational Chemistry, Kluwer Academic Publisher,")
73
[26]. Robert R. G., Shawn C. S., A Chemistry Educators’ Guide to Molecular Modeling, 2007.
[27]. Csizmadia, G. L., 1990, ‘‘Computational adv. in organic chem., molecular str. And reactivity’’, Ed. By Ögretir C. , Csizmadia G. L. , NATO ASI series, Kluwer Academic Publishers., 125. [28]. Dadakdeniz, F., 2007, Halotiyofen Moleküllerinin Elektronik ve Çizgisel Olmayan Optik Özelliklerinin Teorik _ncelenmesi, G.Ü., Fen Bilimleri Enstitüsü. [29]. Doub,L.Richardson,L.M.,Bambas,L.L, Youmans,G.P.,Youmans,A.S.,J.Am.Chem.Soc,80, 2205 (1958). [30]. Rusinov,V.L.,Ulomkii,E.N.,Chupakhin,O.N.Zubairov,M.M.,Kapustin,A.B.,Mitin,N.Zhi ravetskii,M.I.,Vinograd,I.A.,Khim-Farm,Zh.,24,41(1990)(C.A.). [31]. Reader,S.C.J.,Carroll,B.,Robertson,W.R.,LambertA.,Biochem.PHarmacol.,36,1825, (1987). [32]. Kathari,P.J.,Sigh,S.P.,Parmar,S.S.,Stenberg,V.I.HeterocyclicChem.,17,1993(1980). [33]. Lewenstein,M,J.,U.S.P.2, (1954)(C.A.48:13175b). [34]. Gall,M.,Mitarb., HESTER, J.B., RUDZIK, A.D., LAHTI, R.A.: Synthesis and Pharmacology of Novel Anxiolytic Agents Derived from 2-[(Dialkylamino)methyl-4Htriazol- 4-yl]benzophenones and Related Heterocyclic Benzophenones: J.Med.Chem.,19,1057(1976).
![[26]. Robert R. G., Shawn C. S., A Chemistry Educators’ Guide to Molecular Modeling, 2007.](http://slideplayer.biz.tr/slide/9114754/27/images/73/%5B26%5D.+Robert+R.+G.%2C+Shawn+C.+S.%2C+A+Chemistry+Educators%E2%80%99+Guide+to+Molecular+Modeling%2C+2007..jpg "[27]. Csizmadia, G. L., 1990, ‘‘Computational adv. in organic chem., molecular str. And reactivity’’, Ed. By Ögretir C. , Csizmadia G. L. , NATO ASI series, Kluwer Academic. Publishers., 125. [28]. Dadakdeniz, F., 2007, Halotiyofen Moleküllerinin Elektronik ve Çizgisel Olmayan. Optik Özelliklerinin Teorik _ncelenmesi, G.Ü., Fen Bilimleri Enstitüsü. [29]. Doub,L.Richardson,L.M.,Bambas,L.L, Youmans,G.P.,Youmans,A.S.,J.Am.Chem.Soc,80, 2205 (1958). [30]. Rusinov,V.L.,Ulomkii,E.N.,Chupakhin,O.N.Zubairov,M.M.,Kapustin,A.B.,Mitin,N.Zhi. ravetskii,M.I.,Vinograd,I.A.,Khim-Farm,Zh.,24,41(1990)(C.A.). [31]. Reader,S.C.J.,Carroll,B.,Robertson,W.R.,LambertA.,Biochem.PHarmacol.,36,1825, (1987). [32]. Kathari,P.J.,Sigh,S.P.,Parmar,S.S.,Stenberg,V.I.HeterocyclicChem.,17,1993(1980). [33]. Lewenstein,M,J.,U.S.P.2, (1954)(C.A.48:13175b). [34]. Gall,M.,Mitarb., HESTER, J.B., RUDZIK, A.D., LAHTI, R.A.: Synthesis and. Pharmacology of Novel Anxiolytic Agents Derived from 2-[(Dialkylamino)methyl-4Htriazol- 4-yl]benzophenones and Related Heterocyclic Benzophenones: J.Med.Chem.,19,1057(1976).")
74
[35]. Tantwy,A.,Barghash,A.E.M.,Alexandria,J.Pharm.Sci2,50(1988).
[36]. Bonjean,J.,Schunanck,W.,Arch.Pharm 320,554(1987). [37]. Tantwy, A., Barghash, A.E.M., Alexandria, J.Pharm. Sci 2,50 (1988). [38]. Bonjean, J., Schunanck, W., Arch.Pharm 320,554 (1987). [39]. Rusinov,V.L.,Petrov,A.Yu.,Pilicheva,T.L.Chupakhin,O.N.,Kavalev,G.V.,Komina,E.R., Khim-Farm.Zh,20,178(1986) (C.A.106:32968v). [40]. Shah,M.H.,Mhasalkar,M.Y.,Patki,U.M.,Deliwala,C.V.,Sheth,U.K.,J.Pharm.Sci.,58,139 8(1969). [41]. Karyotıkis, N.C. , Anaissie, E.J. , Hachem, R. ,Digmani, M.C. ,Samonis , G., J.Infect. Dis., 168, 131 (1993) [42]. El-Dawy, M.A;, mohsen, A.,Omar, M.E., Ismail, M.A., Hazza, A.A.B.,J.Pharm-Sci- 72,45 (1983). [43]. Greenfild, S.A., SEidel, M.C.,Von Meyer, W.C.,Geroffen ,915 (1970) (C.A.72:100713q). [44]. Dedek,W., Wenzel, K.D., Oberlaender, H., Mothes, B., Maenning, J., Fresenius, 1991, 1.Anal. Chem, 339, 201. [45]. Dubey, S. N. & Yard, B. K., 1991, Synthesis and Characterisation Of Some Bivalent Metal Complexes Of As- Triazine And _ts Schiff Bases, Synth. Reakt. Inorg. Met- Org. Chem., 1299 1311.
![[35]. Tantwy,A.,Barghash,A.E.M.,Alexandria,J.Pharm.Sci2,50(1988).](http://slideplayer.biz.tr/slide/9114754/27/images/74/%5B35%5D.+Tantwy%2CA.%2CBarghash%2CA.E.M.%2CAlexandria%2CJ.Pharm.Sci2%2C50%281988%29..jpg "[36]. Bonjean,J.,Schunanck,W.,Arch.Pharm 320,554(1987). [37]. Tantwy, A., Barghash, A.E.M., Alexandria, J.Pharm. Sci 2,50 (1988). [38]. Bonjean, J., Schunanck, W., Arch.Pharm 320,554 (1987). [39]. Rusinov,V.L.,Petrov,A.Yu.,Pilicheva,T.L.Chupakhin,O.N.,Kavalev,G.V.,Komina,E.R., Khim-Farm.Zh,20,178(1986) (C.A.106:32968v). [40]. Shah,M.H.,Mhasalkar,M.Y.,Patki,U.M.,Deliwala,C.V.,Sheth,U.K.,J.Pharm.Sci.,58,139 8(1969). [41]. Karyotıkis, N.C. , Anaissie, E.J. , Hachem, R. ,Digmani, M.C. ,Samonis , G., J.Infect. Dis., 168, 131 (1993) [42]. El-Dawy, M.A;, mohsen, A.,Omar, M.E., Ismail, M.A., Hazza, A.A.B.,J.Pharm-Sci- 72,45 (1983). [43]. Greenfild, S.A., SEidel, M.C.,Von Meyer, W.C.,Geroffen ,915 (1970) (C.A.72:100713q). [44]. Dedek,W., Wenzel, K.D., Oberlaender, H., Mothes, B., Maenning, J., Fresenius, 1991, 1.Anal. Chem, 339, 201. [45]. Dubey, S. N. & Yard, B. K., 1991, Synthesis and Characterisation Of Some Bivalent. Metal Complexes Of As- Triazine And _ts Schiff Bases, Synth. Reakt. Inorg. Met- Org. Chem.,")
Benzer bir sunumlar

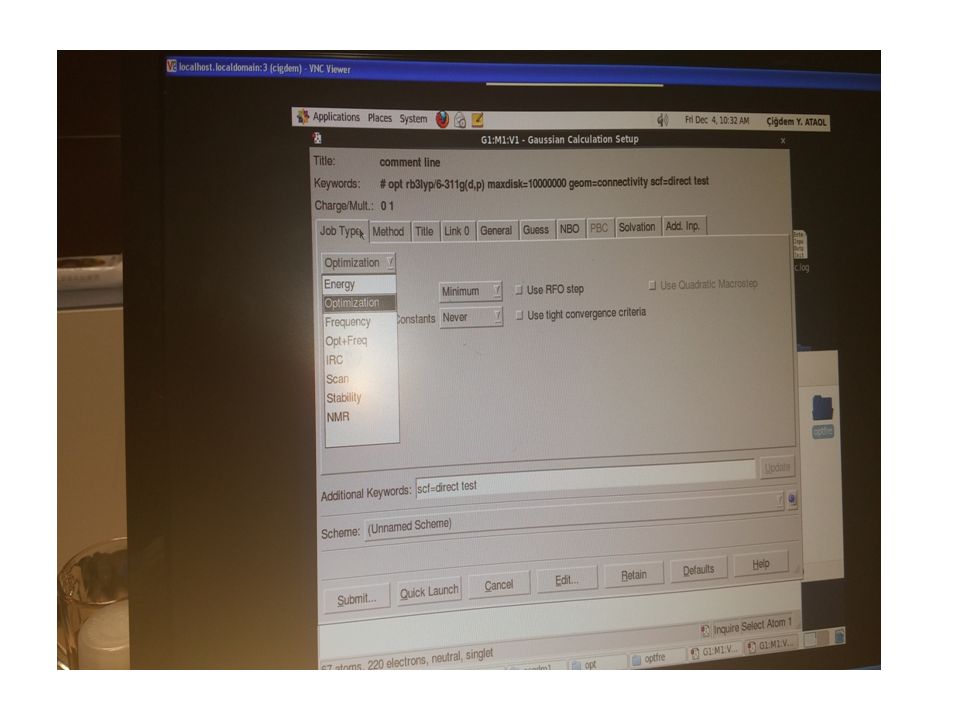
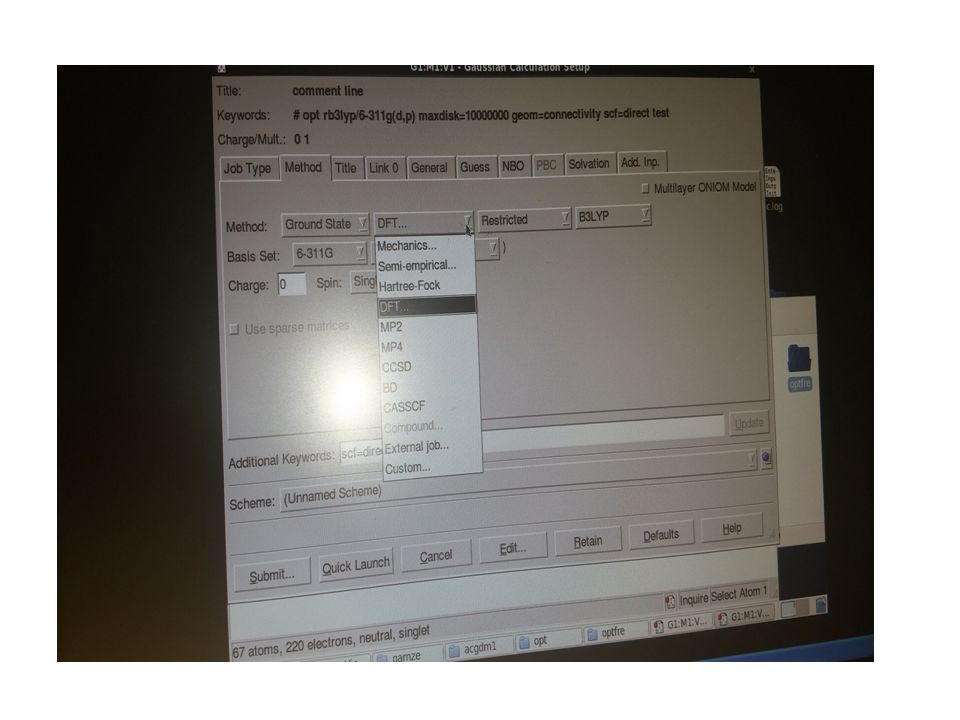
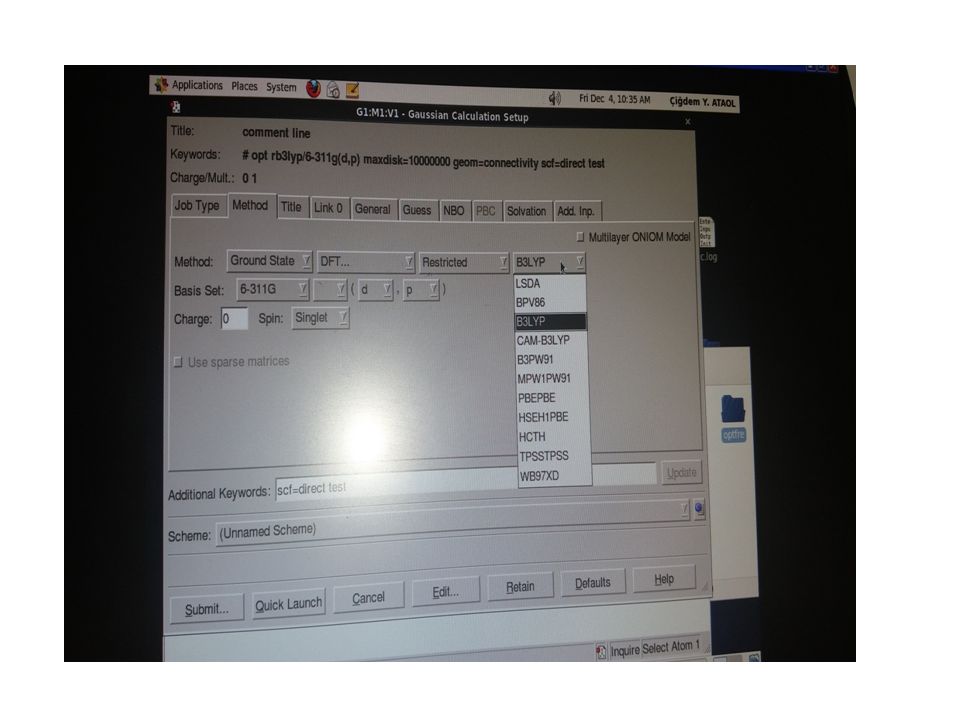
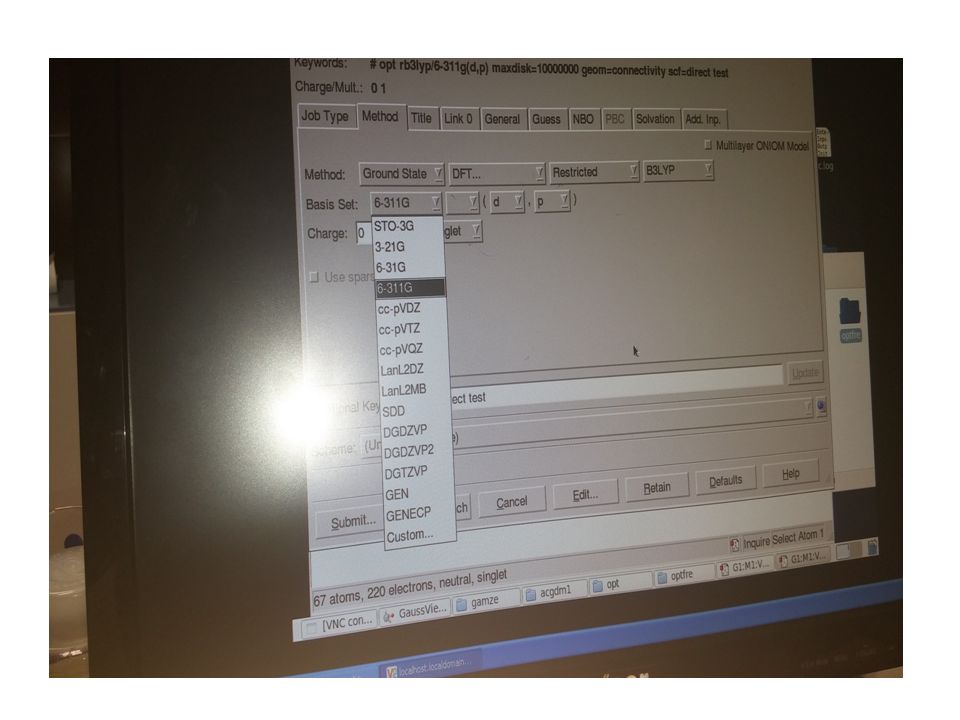

 ile karakterize edilir. Verilen bir elementin tüm atomlarında.>")









 Bağ VBT MOT RAB (kJ/mol)>")







