Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Bu gün Neler öğreneceğiz?
Lizozom mu? O da ne !!!!! Lizozomal enzimler ne zaman aktivite gösterir? Hücrede Lizozomal enzimlerden biri eksik olsa ne olur ki? Depo hastalıkları ne demektir? Peroksizom yapısı ve fonksiyonu nedir? Diğer organellerden farklı mı? Peroksizomlar da mı hastalık nedeni olabilir?

2
Lizozomlar ve Peroksizomlar Hücre içerisinde metabolik işlevlerin
gerçekleştiği yerlerdir. Prof.Dr. Filiz Aydın

3
Lizozom Düzgün bir membranla sınırlı olan veziküler yapılardır,
Hücre içinde bulunan tüm yapıları sindirebilecek özellikte enzimlere sahiptirler, Enzimleri GER’de sentezlenir, Asit ortamda reaksiyon gösteren ~ 60 farklı hidroliktik enzim taşırlar, Enzimler asit karakterlerini ER lümenleri içinde glikozilleşerek kazanırlar,

4
Lizozomal enzimler Cis sisterna içerisinde fosforillenerek, mannoz-6-fosfat artığı ile işaretlenirler, Enzimler aynı zamanda Golgi kompleksinde, Mannoz-6-fosfat artığı, ile diğer ürünlerden ayrılır. Bu artık aynı zamanda enzimlerin farklı bir örtülü vezikül yapısı içinde Golgiden ayrılmasına da olanak sağlar.

5
+ H+ Lizozomal hidrolaz öncüsü Mannoz 6- fosfat Klatrin örtü
Reseptöre bağlı transport ADP + Pi ATP H+ Fosfatın kalkması M6P reseptörüne bağlanma Fosfat eklenmesi Transport vezikülü Olgun lizozomal hidrolaz Geç endozom Reseptörün geri dönüşü Cis Golgi Trans Golgi

6
Golgi sisternalarında iki farklı mannoz -6-fosfat reseptörü bulunmaktadır.
MRP 300 %100 yeterlilikle lizozomal proteinlere bağlanır, MRP 46 daha az etkinlikle enzimlere bağlanarak trans Golgi’den veziküler yapı halinde ayrılmalara izin verir. Lizozomal enzimler hangi tip veziküler yapılar halinde Golgi’yi terk eder?

7
Primer – sekonder lizozom nedir?
Golgi’yi terk eden veziküler yapılar Primer Lizozom olarak isimlendirilir. Mannoz-6-fosfat artıkları defosforilasyon ile enzimden ayrılır. Primer lizozom içeriğini endozom olarak isimlenen bir vezikül içine akıtır. Primer lizozom ve endozomun füzyonu ile meydana gelen yapı artık Sekonder lizozom (geç endozom) olarak isimlendirilmektedir
 olarak isimlendirilmektedir.")
8
Primer lizozom Füzyon sonucu şekillenmiş sekonder lizozom Golgi apereyi

9
Sekonder Lizozomlar sindirecekleri materyalin
niteliğine göre iki farklı şekilde adlandırılır ! Sekonder Lizozom Heterofagozom (heterofajik vakuol-fagolizozom). Otofagozom ( otofajik vakuol-sitolizozom sitozom). Heterofagozom ile otofagozom arasında ne fark vardır?
. Otofagozom ( otofajik vakuol-sitolizozom sitozom). Heterofagozom ile otofagozom arasında ne fark vardır")
10
Plazma membranı Fagozom bakteri Primer lizozom fagositoz Primer lizozom Endositoz Sekonder lizozom Erken endozom Geç endozom Primer lizozom Mitokondri ER Otofagozom Otofaji

11
Sitozol endositoz fagositoz otofaji Golgi apereyi endozom
Endoplazmik Retikulum Golgi apereyi endozom Hidrolitik enzim içeren transport vezikül fagozom organel otofagozom endolizozom fagolizozom otofagolizozom Sitozol

12
Peroksizom fragment Mitokondri fragment Lizozom

13
Lizozomların hücrede üç önemli fonksiyonu vardır-1;
Büyük besin partiküllerinin sindirimi (Bakterilerin de sindiriminde olduğu gibi), Endositoz ile hücreye alınan moleküler materyallerin modifikasyonu ve depolanması, Yaşlanan ya da hasarlı organellerin yıkımı.
, Endositoz ile hücreye alınan moleküler materyallerin. modifikasyonu ve depolanması, Yaşlanan ya da hasarlı organellerin yıkımı.")
14
Lizozomal enzimler aktivitelerini pH5 ortamında gösterebilirler.
Lizozomal membranda bulunan ATPaz, ATP’yi parçalar ve açığa çıkan enerji ile H+’ yi lizozom içerisine pompalar. Lizozomal membranlar yüksek derecede glikozilleşme ile enzim sindiriminden kendilerini koruyabilir. Lizozom içindeki makromoleküllerin sindirimi sonucu açığa çıkan küçük moleküller ise membrandaki transport proteinler aracılığı ile sitozole gönderilir. Sindirilemeyen materyaller ise artık cisimler olarak hücre içinde kalabilirler. Kalp kası ve nöronlarda artık cisimler-lipofuskin / yaşlılık pigmenti

15
+ Sitazol Glikozillenerek kendi enzimlerinden korunma sağlanır pH~7.2
Pi ATP ADP H-ATPaz Lizozomlardaki proteazlar,glikozidazlar,nükleazlar,fosfatazlar,lipazlar v.b enzimler asidik pH (~5) da etkinlik gösterir.
 da etkinlik gösterir.")
16
Lizozomal enzimler ve fonksiyonları
Nükleik asit degradasyonu Asit RNAase ve asit DNAase RNA ve DNA Karbonhidrat degredasyonu - galaktosidaz -Glukosida Lizozim Galaktosidler Glukojen Bakteri hücre duvarı Mukopolisakkarid degradasyonu -Glukorinidaz Hiyaluronidaz Mukopolisakkaridler Hiyaluronat ve kondraatin sülfat Protein degradasyonu Katepsin Kollojenaz Proteinler Kollojen Fosfat Asit fosfataz Asit fosfodiesteraz Fosfomonoesterler Oligonukleotidler Sulfat Arilsülfetaz Organik sülfatlar

17
Lizozomal enzimlerin görevi organizmayı hastalıklara karşı korumaktadır.
Eritrositlerde lizozomal yapılar bulunmazken özellikle bazı hücrelerin çok sayıda bu organele sahip oldukları görülmektedir. Bu hücreler: ??? Patolojik şartlarda membran geçirgenliğinin artması ile enzimler dışarı çıkarak hastalıklara neden olabilir. Lökosit lizozomlarında var olan baz yapılı proteinler Mast hücreleri aracılığı ile kılcal damar endotelindeki geçirgenliği etkiler. Kan plazma ve hücreleri dokuya geçerek ödem ve iltihaplanmaya neden olur.

18
Kronik romatoid artrit
Lokositler eklem sıvısında toplanan Antijen –antikor komplekslerini fagosite eder, Lizozomal membranların geçirgenliği bu fagositasyona bağlı olarak artar enzimler açığa çıkarak eklem aralığında toplanır, SONUÇ: eklem kıkırdaklarının yapısı bozulur. Tedavi:Membran geçirgenliğini normale döndüren bileşikler (kortizon- hidrokortizon) tedavi amaçlı olarak kullanılır.
 tedavi amaçlı olarak kullanılır.")
19
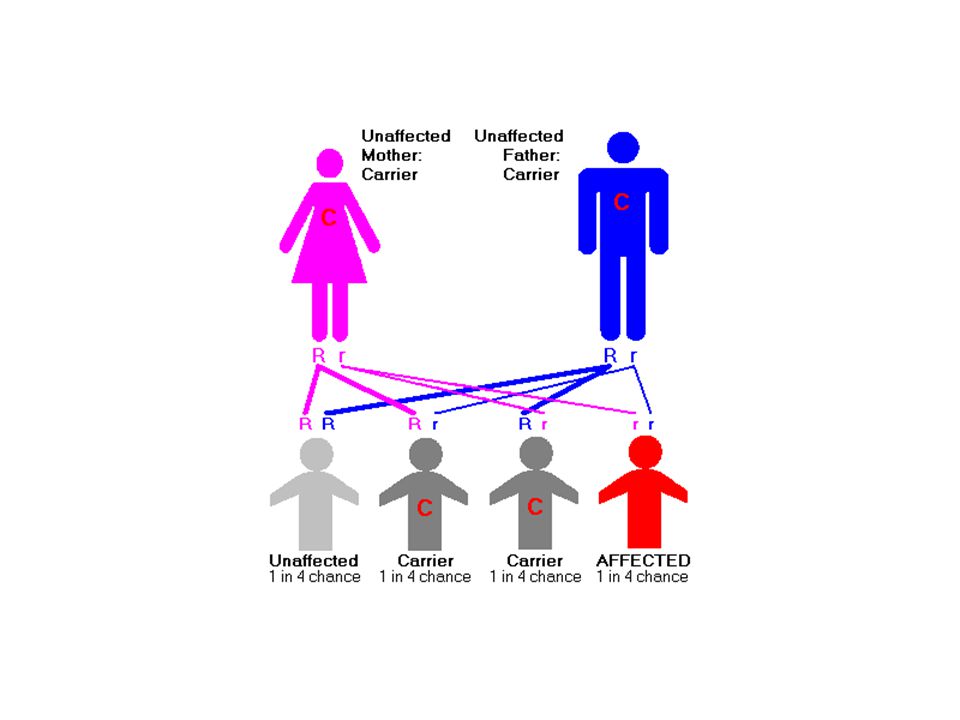
Lizozomal enzim eksiklikleri bazı metabolik
hastalıklara neden olur.

20
Tay-sachs hastalığı (TSD)
Sinir hücrelerinde çok bulunan gangliozidlerin (glikolipid) parçalanmasından sorumlu, heksozaminidaz A enzimi eksikliği ile, oluşur. Resesif kalıtım özelliği gösteren bir hastalıktır
 parçalanmasından sorumlu, heksozaminidaz A enzimi eksikliği ile, oluşur. Resesif kalıtım özelliği gösteren bir hastalıktır.")
22
TDS-Nörolojik semptomlara göre sınıflandırılırlar
1-İnfantil TSD; İlk altı ayda kadar normal olarak TSD’ li bebeklerde görülmeye başlar. Genellikle 3 yaş öncesi ölümle sonuçlanır 2- Juvenil TSD; çok ender olarak yaş çocuklarda görülür yaşlarda ölüm ile sonuçlanır 3- Adult TSD ; Nadiren görülür, ilaç tedavisi ile semptomlar önlenebilir

23
Lizozomal enzimlerin eksikliğine bağlı hastalıklar
Eksik olan enzim Etkilenen hücre Etkilenen doku GAUCHER -d-glikosidaz Makrofajlar/glikolipid birikimi Karaciğer ve dalak METAKROMATİK LÖKODİSTROFİ Sülfataz Dermatan sülfat birikimi Çeşitli dokular HURLER L- iduronidaz Fibroblast ve osteoblastlarda Dermatan sülfat birikimi İskelet ve sinir sistemi TAY-SACHS Heksoaminidaz Sinir hücreleri Glikolipid birikimi Sinir sistemi SANFİLİPPO SENDROMU Heparin sülfat sülfamidaz Fibroblastlarda Heparan sülfat birikimi İskelet sistemi

24
GAUCHER HASTALIĞI TİP 1 İlk başta hastada ciddi KC ve dalak tutulumu vardı. Splenoktomi sonrası hasta. KC abdominal boşluğu dolduruyor. Uzun kemik tutulumundan dolayı bacaklar çanak görüntüsü almış Hızlı progresif hastalık belirtileri yaygın olarak çocuklarda görülür.

25
Lizozomal enzim bozukluğuna bağlı olan bu hastalıklar;
DEPO hastalıkları olarak isimlendirilir. Nedeni; Lizozomal enzim genlerindeki mutasyonlar, Mannoz 6-fosfat tanıma sinyalini oluşturan gen mutasyonu.

26
+ H+ Lizozomal hidrolaz öncüsü Mannoz 6- fosfat Klatrin örtü
Reseptöre bağlı transport ADP + Pi ATP H+ Fosfatın kalkması M6P reseptörüne bağlanma Fosfat eklenmesi Transport vezikülü Olgun lizozomal hidrolaz Geç endozom Reseptörün geri dönüşü Cis Golgi Trans Golgi

27
Hurler sendromu I-hücre hastalığı Normal hücre Enzimsiz Golgi
Enzimsiz lizozom I-hücre hastalığı Normal hücre Ekli olmayan enzimli Golgi Hücreden ayrılan vezikül Golgi aygıtı Enzim Eklenme Test tüpünde hücreye verilen ekli enzimler Enzimli lizozom Boş lizozom Alınmaz Lizozom Enzim eksik

28
Peroksizomlar Peroksizomlar 40 farklı enzim taşıyan membranla sınırlı
organellerdir. Bölünerek çoğalabilen organel tüm ökaryotik hücrelerde bulunmakla beraber özellikle omurgalılarda; Karaciğer ve böbrek hücrelerinde bol miktarda bulunurlar. Önemli enzimleri; Urat oksidaz D-amino asid oksidaz Katalaz’ dır .

30
Peroksizomal enzimler nerede sentezlenir?
Peroksizomal enzimler lizozomal enzimlerin aksine SİTOZOL’de sentez edilirler. Organele ait membran proteinleri de sitozolde sentezlenmektedir Peroksizomal proteinlerin karboksi terminal uçlarında üç amino asitlik bir dizi sinyal özelliği göstermektedir. Peroksizoma yönlendirici bu sinyal enzimlerin organele taşınmasında önemlidir. ENZİMLER; Organel membranında yer alan, membran proteinleri tarafından tanınan transport sinyal aracılığı ile organel içine alınır.

31
Bölünerek çoğalabilirler
bölünme Bölünerek çoğalabilirler

32
Oksidatif reaksiyon; RH2 + O2 R + H2O2 Bu tip reaksiyonlar;
Kana geçen toksik maddelerin detoksifikasyonunu sağlarlar. O2 ‘ yi kullanarak Oksidatif reaksiyonları yapabilirler. Organik moleküllerden hidrojeni uzaklaştırmak için moleküler oksijeni kullanabilirler.

33
Peroksidatif reaksiyon; H2O2 +R’H2 R’ + 2H2O
Katalaz enzimi, oksidatif reaksiyon sonucu açığa çıkan H2O2‘ti Fenoller Formik asit Formaldehid Alkol’ü içeren Çeşitli maddelerin detoksifikasyonunda kullanır.

34
Peroksizomal hastalıklar
İnsanda çeşitli peroksizomal hastalıklar bilinmektedir. Tümü otozomal resesif kalıtsal özellik göstermektedir. Zellweger sendromu Adrenolökodistrofi İnfantil refsum hastalığı İnfantil refsum hastalığı vücutta peroksizomların azalması veya yokluğu ile karakterize bir hastalıktır. Plazma ve dokularda fitanik asit birikir. Bu hastalık beyinde sinir fibrilerinin ve myelin kılıfın gelişimini etkileyen lökodistrofi diye adlandırılan genetik hastalıklar grubundan bir hastalıktır. Adrenolökodistrofi 2 ayrı genetik hastalıktan oluşur. Bunlar X-linked ALD ve neonatal ALD’dir. Bu iki hastalık değişik derecelerde adrenal tutulum ve demyelinizasyon ile karakterizedir. Serum ve dokularda uzun zincirli yağ asitleri birikir.

35
Zellweger sendromu Kalıtsal bir hastalıktır. Sebep;
Peroxisome assembly factor-1 olarak isimlenen peroksizomal integral membran proteini’ni kodlayan gen üzerindeki mutasyondur. Sonuç; İçi boş peroksizomların varlığı, peroksizomal yetersizlikler, Beyin, Karaciğer ve Böbrekte ciddi anomaliler, Hastaların doğum sonrası kaybı.

36
Neler öğrendik ! Lizozom ve peroksizomun *yapısı, *fonksiyonu ve
*özellikleri, 2. Depo hastalıkları nedir, neden meydana gelir,

37
ÖDEV Ailesel hiperkolesterolomi nedir? Neden meydana gelir?
Kaynak kitaplar: Tıbbi genetik ve İç hastalıkları

Benzer bir sunumlar





>")














