Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Redefining Alzheimer’s Disease
Hakan Gürvit Istanbul University Istanbul Faculty of Medicine Department of Neurology Behavioral Neurology and Movement Disorders Unit

2
Auguste Deter Alzheimer A. Allgem Z Psychiatr Psych- Gerich Med 1907
Auguste D 1850 Mayıs’ında doğmuştu. 1890’ların sonlarına doğru bellek ve dil bozuklukları şeklinde tanımlanan demans semptomları geliştirmeye başladı. Kısa zamanda ciddi psikotik özelllikler ve ajitasyon eklendi ve Kasım 1901’de , Alois Alzheimer tarafından muayene edileceği Frankfurt Akıl Hastanesi’ne getirildi, 8 Nisan 1906’da bu hastanede öldü. 3 Kasım 1906’da, Tübingen’deki Güneydoğu Alman Psikiyatri Derneği’nin 37. Toplantısı’nda Auguste D:’nin klinik ve nöropatolojik bulgularını "Über einen eigenartigen, schweren Erkrankungsprozess der Hirnrinde" isimli 11. bildiriyle sundu. Hastanın yaşının genç olması dolayısıyla hastalık bir pre-senil demans biçimi olarak değerlendirilecektir. Beynin nöropatolojik incelemesinde serebral korteksin normale göre incelmiş olduğunu gördü. Ayrıca, daha sonra amiloid plaklar olarak adlandırılacak miliyer odakları ve yine daha sonra nörofibriler yumaklar adını alacak intraselüler fibril birikintilerini tanımladı. Alzheimer A. Allgem Z Psychiatr Psych- Gerich Med 1907

3
Alzheimer’s Disease is a Rare Mental Disorder
Emil Kraepelin. Psychiatrie: ein Lehrbuch für Studierente und Ärzte. (Leipzig, 1910) Alzheimer’s disease is a presenile dementia
 Alzheimer’s disease is a presenile dementia.")
4
AD is the Most Prevalent Dementia
B.E. Tomlinson, G. Blessed and M. Roth Observations on the brains of non-demented old people. Journal of Neurological Sciences (1968) 7, Observations on the brains of demented old people. Journal of the Neurological Sciences (1970) 11, “Senile dementia is not arteriosclerotic but is associated with AD-like changes.” R. Katzman The prevalence and malignancy of Alzheimer’s disease. A major killer. Arch Neurol 1976;33:217–8. “Senile dementia is identical with Alzheimer’s disease.”
 7, Observations on the brains of demented old people. Journal of the Neurological Sciences (1970) 11, Senile dementia is not arteriosclerotic but is associated with AD-like changes. R. Katzman. The prevalence and malignancy of Alzheimer’s disease. A major killer. Arch Neurol 1976;33:217–8. Senile dementia is identical with Alzheimer’s disease.")
5
Alzheimer Type Dementia
Multiple cognitive deficits with an amnestic core, disrupting ADL’s NINCDS-ADRDA (1984) Probable AD (PRAD) DSM-IV (1994) AD-type dementia (DAT) The clinician is excluded from the definite diagnosis of AD, which is granted to the pathologist as the exclusive right.
 Probable AD (PRAD) DSM-IV (1994) AD-type dementia (DAT) The clinician is excluded from the definite diagnosis of AD, which is granted to the pathologist as the exclusive right.")
6
Pre-Dementia Memory Impairment
BSF: Forgetful elderly, who do not progress into dementia MCD (ICD-10), MNCD (DSM-IV), AACD (IPA), CIND (CSHA) Age-associated Memory Impairment (AAMI)2 A person aged over 50 with subjective memory complaints and a formal memory performance which is 1SD lower than that of the normative mean of the young adults2 Mild cognitive impairment (MCI)3 Objective evidence of memory impairment as compared to age norms, no functional impairment3 1Kral 1962 2NIA 1986 3Petersen et al. 2001
, MNCD (DSM-IV), AACD (IPA), CIND (CSHA) Age-associated Memory Impairment (AAMI)2. A person aged over 50 with subjective memory complaints and a formal memory performance which is 1SD lower than that of the normative mean of the young adults2. Mild cognitive impairment (MCI)3. Objective evidence of memory impairment as compared to age norms, no functional impairment3. 1Kral NIA Petersen et al")
7
Memory Impairment Continuum
Healthy Aging Memory Impairment Continuum None Age- Associated Memory Impairment (AAMI) As compared to young adults Mild Cognitive Impairment (MCI) Isolated, no functional impairment Prominent among multiple cognitive deficits, with functional impact Mild Dementia Severe with intact basic ADL’s Moderate Dementia Severe Dementia Severe, totally dependent 10 20 30 40 50 60 70 80 90
 As compared to. young adults. Mild. Cognitive. Impairment. (MCI) Isolated, no functional impairment. Prominent among multiple cognitive. deficits, with functional impact. Mild. Dementia. Severe with intact basic ADL’s. Moderate. Dementia. Severe. Dementia. Severe, totally dependent")
8
Spatio-Temporal Distribution of NFT Pathology in AD
Tauoism: High clinical correlation with NFT distribution Space M-M Mesulam 2000 Low Limbic (Normal/ AAMI) + High Limbic (MCI) ++ + Low Neocortical (variable AD) +++ ++ + High Neocortical (severe AD) ++++ +++ ++ + Time Entorhinal Limbic Paralimbic Prefrontal STS, iPL Unimodal Ass Primary S-M Braak-Braak. NeurAging 1995 I-II III-IV V VI
 + High Limbic. (MCI) ++ + Low. Neocortical. (variable AD) High Neocortical. (severe AD) Time. Entorhinal. Limbic. Paralimbic. Prefrontal. STS, iPL. Unimodal Ass. Primary S-M. Braak-Braak. NeurAging I-II. III-IV. V. VI.")
9
Familial Alzheimer’s Disease
Chr 21. APP (Goate, Hardy, Nature 1991) Chr 14. PSEN1 (Sherrington, St. George-Hyslop, Nature 1995) Chr 1. PSEN2 (Levy-Lahad, Schellenberg, Science 1995) Chr 19. APOE polymorphism (Strittmatter, Lancet 1996)
 Chr 14. PSEN1 (Sherrington, St. George-Hyslop, Nature 1995) Chr 1. PSEN2 (Levy-Lahad, Schellenberg, Science 1995) Chr 19. APOE polymorphism (Strittmatter, Lancet 1996)")
10
Baptism : Amyloid Cascade Hypothesis
The prime mover of AD neuropathology is Aβ peptide. All the genetic causes of AD, whether Mendelian mutations or susceptibility polymorphisms, affect either by increasing Aβ levels or decreasing its clearence from the brain parenchyma Tau gene (MAPT) mutations cause FTD, but never AD Hardy & Allsop, Trends Pharmacol Sci 1991
 mutations cause FTD, but never AD. Hardy & Allsop, Trends Pharmacol Sci")
11
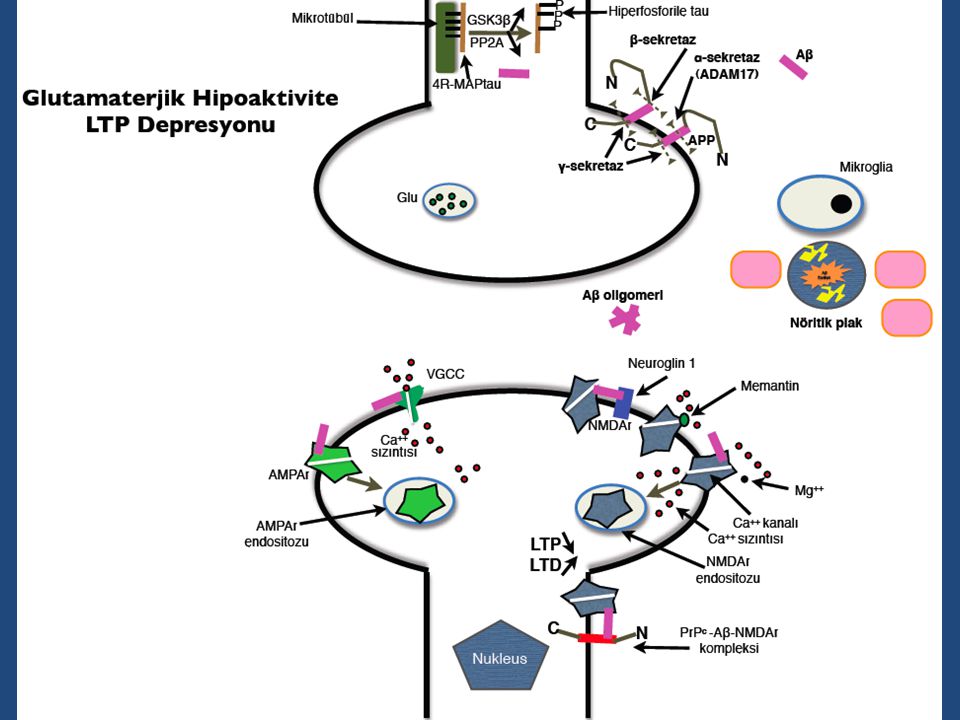
Lewy cisimcikleri, TDP-43, FUS)
Fagozom /lizozomlar İntrasitoplazmik inklüzyon (NFY’ler, Lewy cisimcikleri, TDP-43, FUS)
")
12
Nörofibriler Yumak İntraselüler birikim
NFY’ler ilk kez 1977’de izole edilmiş (Cleveland et al.) olan tau proteinine immünoreaktif (Brion et al. 1985) NFY’nin temel bileşeni fosforillenmiş proteinidir (Grundke-İqbal et al. 1986) 17.kromozomda kodlanan mikrotübül asosiye proteinlerden (MAPT) biri (Hutton et al. 1998) İşlevi aksonal transport (kargo vezikülleri: yenilenme ve sinaptik aktivite için gerekli enzimler) ve sitoskeletal bütünlük
 olan tau proteinine immünoreaktif (Brion et al. 1985) NFY’nin temel bileşeni fosforillenmiş proteinidir (Grundke-İqbal et al. 1986) 17.kromozomda kodlanan mikrotübül asosiye proteinlerden (MAPT) biri (Hutton et al. 1998) İşlevi aksonal transport (kargo vezikülleri: yenilenme ve sinaptik aktivite için gerekli enzimler) ve sitoskeletal bütünlük.")
13
MAP-τ’nun NFY’ye Evrimi

14
Amiloid Plaklar AP’ler ekstraselüler birikim
Amiloid yükü hastalık şiddetiyle korele değil Amiloid maddelere yüksek affiniteli Kongo kırmızısı ile boyanırlar (Divry 1927) Temel bileşenleri A peptidi (Glenner ve Wong 1984) A 21. kromozomda kodlanan bir transmembran protein APP’nin çözülemeyen fragmanı (Hardy 1991) Gevşek plaklarda non-fibriler (granüler) Aβ42 Sert plaklarda fibriler Aβ42 ve Aβ40 birlikte
 Temel bileşenleri A peptidi (Glenner ve Wong 1984) A 21. kromozomda kodlanan bir transmembran protein APP’nin çözülemeyen fragmanı (Hardy 1991) Gevşek plaklarda non-fibriler (granüler) Aβ42. Sert plaklarda fibriler Aβ42 ve Aβ40 birlikte.")
15
Çözülebilir Oligomerik vs. Çözülemeyen Fibriler Aβ
Fibrilizasyon amiloid yapısının ilk elektron mikroskopik belirlenimlerinden beri nörodejenerasyonla ilişkilendirilmiştir (Cohen and Calkins, 1959; Kidd, 1964; Terry et al.,1964). Yakınlarda çözülebilir, oligomerik yapıların güçlü nörotoksik aktiviteleri gösterildi. Ekstraselüler amiloid depozitleri, intraselüler aggregatlar, (örn, aggresomlar) (Kopito, 2000) hastalık seyrinde geç evre özellikleri olabilir; çözülebilir asambleler nöronal disfonksiyona yolaçacak şekilde daha erken dönemde etki ediyor olabilirler (Bucciantini et al., 2002). Aβ fibrillerinin in vitro analizi bir dizi oligomerik ara yapılar ortaya koymuştur: Özellikle, protofibriller ve Aβ’dan türeyen çözülebilir ligandlar (ADDL’ler).
. Yakınlarda çözülebilir, oligomerik yapıların güçlü nörotoksik aktiviteleri gösterildi. Ekstraselüler amiloid depozitleri, intraselüler aggregatlar, (örn, aggresomlar) (Kopito, 2000) hastalık seyrinde geç evre özellikleri olabilir; çözülebilir asambleler nöronal disfonksiyona yolaçacak şekilde daha erken dönemde etki ediyor olabilirler (Bucciantini et al., 2002). Aβ fibrillerinin in vitro analizi bir dizi oligomerik ara yapılar ortaya koymuştur: Özellikle, protofibriller ve Aβ’dan türeyen çözülebilir ligandlar (ADDL’ler).")
16
Toksik C N C N QC LMW-Aβ ADDL Gevşek AP Aβ protofibrilleri
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA α-sekretaz 49/50 1 ε ζ γ Non-amiloidojenik Proteoliz AICD C AICD p3 sAPPα N Gen ifadesi 770 687 1 717/718 QC γ-sekretaz kompleksi 1 40/42 β-sekretaz DAEFRHDSGYE…………A Aβ Nukleus 4kDa 711/713 Amiloidojenik Proteoliz 720/721 C AICD Aβ sAPPβ N Gen ifadesi 770 671 1 8-20kDa 723 700 LMW-Aβ ADDL NICD Gevşek AP Lizozomal Aβ(pE)3-42 Aβ(pE)11-42 Membran Dimer Tetramer Proteozomal APO E α2M Nep IDE Plasmin Psen 1/2 Pen-2 Nicastrin Aph1 A/B Mikroglial fagositoz NOTCH kDa Nöronal proteoliz Astrositik otofaji Aβ protofibrilleri α→β Aβ*56 - Dodekamer Globulomer Sferoid İntranöronal agrezomlar >100 kDa Fibriler Aβ Sert AP Toksik Jacobsen et al. PNAS 2008
3-42. Aβ(pE) Membran. Dimer. Tetramer. Proteozomal. APO E. α2M. Nep. IDE. Plasmin. Psen 1/2. Pen-2. Nicastrin. Aph1 A/B. Mikroglial. fagositoz. NOTCH kDa. Nöronal. proteoliz. Astrositik. otofaji. Aβ protofibrilleri. α→β. Aβ*56 - Dodekamer. Globulomer. Sferoid. İntranöronal. agrezomlar. >100 kDa. Fibriler Aβ. Sert AP. Toksik. Jacobsen et al. PNAS")
18
A Decade of Glory 1993-2002 Tacrin 1993 Donepezil 1996
Rivastigmine 1997 Galantamine 2001 Memantin 2003

19
A Decade of ACH-based Disappointments
Phase III Failures AN 1792 (2002) – Phase II, Active immunisation Alzhemed (2007) – Phase III, Aβ anti-oligomerisator Flurizan (2009) – Phase III, GSM Dimebon (2010) – Faz III, Mitochondrial stabilisator Semagacestate (2010) – Phase III, GSI TTP488 (PF ) (2011) – Phase II, RAGE iinhibitor Ponezumab (2011) – Phase II, Passive immunisation Bapineuzumab (2012) – Phase III, Passive immunisation Despite the total clearence of AP’s in 7/8 autopsy cases from AN1792, their last recorded MMSE scores before death were 0.
 – Phase II, Active immunisation. Alzhemed (2007) – Phase III, Aβ anti-oligomerisator. Flurizan (2009) – Phase III, GSM. Dimebon (2010) – Faz III, Mitochondrial stabilisator. Semagacestate (2010) – Phase III, GSI. TTP488 (PF ) (2011) – Phase II, RAGE iinhibitor. Ponezumab (2011) – Phase II, Passive immunisation. Bapineuzumab (2012) – Phase III, Passive immunisation. Despite the total clearence of AP’s in 7/8 autopsy cases from AN1792, their last recorded MMSE scores before death were 0.")
20
Bapineuzumab Phase II – PIB-PET
Rinne et al. Lancet Neurol 2010

21
In-vivo Amyloid Load: PIB-PET
Klunk, Engler, Nordberg et al. Ann Neurol 2004

22
2-year Follow-up of the Original Study
Re-scanning of the 16 AD subjects Despite 20% decrease in the glucose metabolism with FDG-PET, no substantial increse in amyloid load with PIB-PET Engler H et al. Brain 2006 Klunk et al. Brain 2006

23
PIB-PET in Healthy Aging, MCI and AD
Pike et al. Brain 2007 31 AD, 33 MCI, 32HA. PIB retention in 97% of AD, 61% of MCI, 22% of HA subjects.

24
Longitudinal Studies with Amyloid Imaging in MCI
21 MCI; Karolinska (Forsberg et al., Neurobiol Aging 2008) 10/21 PIB+ aMCI; conversion to AD in 8 months 7/10 in PIB+ and 0/11 in PIB- groups 26 MCI; Pittsburgh (Wolk et al, Ann Neurol 2009) 13/26 PIB+; 5/13 PIB+ and 0/13 PIB- convert to AD in 21.2±16 mo. 31 aMCI; London (Okello et al., Neurology 2009) 14/17 PIB+ (1/14 PIB-) convert to AD in 3 years 8/14 in 1 year: all of whom are ε4 carriers with higher PIB load 65 MCI; Heidelberg, Australia (Villemagne et al., Ann Neurol 2011) 35/45 PIB+ convert in 2 years (1/20 PIB- [4/20 to non-AD])
 10/21 PIB+ aMCI; conversion to AD in 8 months 7/10 in PIB+ and 0/11 in PIB- groups. 26 MCI; Pittsburgh (Wolk et al, Ann Neurol 2009) 13/26 PIB+; 5/13 PIB+ and 0/13 PIB- convert to AD in 21.2±16 mo. 31 aMCI; London (Okello et al., Neurology 2009) 14/17 PIB+ (1/14 PIB-) convert to AD in 3 years. 8/14 in 1 year: all of whom are ε4 carriers with higher PIB load. 65 MCI; Heidelberg, Australia (Villemagne et al., Ann Neurol 2011) 35/45 PIB+ convert in 2 years (1/20 PIB- [4/20 to non-AD])")
25
Longitudinal Studies with Amyloid Imaging in Healthy Control Subjects
106 HS; Heidelberg, Australia (Villemagne et al., Ann Neurol 2011) 33/106 PIB+; 5/33 in 20 mo., 11/33 in 36 mo. convert to MCI/AD (1/73 PIB-) Binding is predominantly prefrontal and precuenus 146 HS; Wash Uni, St. Louis (Vlassenko et al., Ann Neurol 2011) In 2.5 years 17/21 PIB+ subjects become high load-carriers (8%/year increase) No reversion into PIB- state 10/125 PIB- become PIB+ (%3.1/year conversion rate) 7%/year in ε4 carriers
 33/106 PIB+; 5/33 in 20 mo., 11/33 in 36 mo. convert to MCI/AD (1/73 PIB-) Binding is predominantly prefrontal and precuenus. 146 HS; Wash Uni, St. Louis (Vlassenko et al., Ann Neurol 2011) In 2.5 years 17/21 PIB+ subjects become high load-carriers (8%/year increase) No reversion into PIB- state. 10/125 PIB- become PIB+ (%3.1/year conversion rate) 7%/year in ε4 carriers.")
26
PIB Load in HS Villemagne et al., Ann Neurol 2011

27
Beynin Olağan (“Default”) İşlevi Hipotezi
Nicel PET ile PCC ve vACC’yi de içeren bir dizi beyin bölgesinde gözleri kapalı, istirahat halindeki bireylerde süregiden harici ipuçlu görevlerin performansı sırasında askıya alınan aktivite “These decreases suggest the existence of an organized, baseline default mode of brain function that is suspended during specific goal-directed behaviors.” Raichle et al. PNAS 2001

28
DMN Aktivitesi AH’yi Sağlıklı Yaşlanmadan Ayırır
A: Sağlıklı yaşlanma B: Erken AH Duyarlılık: %85 Özgüllük: %77 Grecius et al. PNAS 2004

29
Amiloid Yükü ve DMN Aktivitesi
AH’liler ve PIB+ normal kognisyonlu bireyler PIB- normal kognisyonlu bireylere göre benzer şekilde anlamlı düzeyde DMN hipoaktivitesi gösterirler. AH : 35 PIB+ : 20 PIB- : 48 Sheline et al. Biol Psychiatry 2010

30
APOE Polimorfizm ve DMN
DMN aktivitesi ε4+/PIB- normal bireylerde, ε4-/PIB- bireylere kıyasla, PIB+ bireyler ve AH’lilere benzer tarzda değişir. Sheline et al. J Neurosci 2010

31
Clinical Progression is Driven by Tau but not by Amyloid Deposition
21 HS, 32 aMCI, 8 AD, 2 years; Mayo, USA (Jack et al., Brain 2009) Increase in PIB load: HS>MCI>AD Ventriculer enlargement: HS<MCI<AD Cognitive measures correlate with VE bot not with PIB load The presence of amyloid load is not sufficient for cognitive impairment; it seems to be driven by neurodegeneration 137 MCI; Malmö, Sweden (Buchhave et al., 2012) 72/137 convert to AD in 9.2 years (21 to non-AD) Early converters (EC: <5 years) and late converters (LC: >5 years) have equal amount of CSF-Aβ42 CSF-pTau-tTau: EC>LC
 Increase in PIB load: HS>MCI>AD. Ventriculer enlargement: HS<MCI<AD. Cognitive measures correlate with VE bot not with PIB load. The presence of amyloid load is not sufficient for cognitive impairment; it seems to be driven by neurodegeneration. 137 MCI; Malmö, Sweden (Buchhave et al., 2012) 72/137 convert to AD in 9.2 years (21 to non-AD) Early converters (EC: <5 years) and late converters (LC: >5 years) have equal amount of CSF-Aβ42. CSF-pTau-tTau: EC>LC.")
32
Amyloidosis and Neurodegeneration are Separate Processes
819 HS, MCI and AD, 59 ADNI centers, 2 years (Lo et al., Arch Neurol 2011) CSF-Aβ42 decrease: HC>MCI>AD FDG-PET metabolism and Hipp Atrophy: HC< MCI < AD Amyloid load is an earlier event as compared to hypometabolism and atrophy 110 MCI, low CSF-Aβ42; Amsterdam (van Rossum et al., Neurology 2012) 63/110 AD converters in 2.2 years. Conversion HR’s: CSF-t-Tau 2.3, CSF-pTau 3.5, MRI-Hipp atrophy 2.5, t-Tau+HA 7.3 Neuronal damage markers in cerebral amyloidosis are the predictors of conversion
 CSF-Aβ42 decrease: HC>MCI>AD. FDG-PET metabolism and Hipp Atrophy: HC< MCI < AD. Amyloid load is an earlier event as compared to hypometabolism and atrophy. 110 MCI, low CSF-Aβ42; Amsterdam (van Rossum et al., Neurology 2012) 63/110 AD converters in 2.2 years. Conversion HR’s: CSF-t-Tau 2.3, CSF-pTau 3.5, MRI-Hipp atrophy 2.5, t-Tau+HA 7.3. Neuronal damage markers in cerebral amyloidosis are the predictors of conversion.")
33
Interim Conclusions Aβ plaque accumulation is a slow process and seems to terminate before cerebral atrophy and cognitive impairment It is age-related >64 years: %20; >85 years: %85 Amyloid imaging: 2/3 MCI and 1/3 HS positive Amyloid positivity is the predictor of conversion from HS to MCI and from MCI to AD Amyloid positivity is diagnostic in atypical AD presentations Amiloid negativity is a marker of non-AD dementia

34
NIA-AA Instead of NINCDS-ADRDA
Biomarkers (BM) Molecular CSF Aβ and tau PET amyloid imaging Structural imaging Hippokampal volumetry Cortical thickness Metabolic imaging FDG-PET Aβ accumulation markers CSF Aβ and amyloid imaging Neurodegeneration markers CSF tau and structural/metabolic imaging Pre-clinical AD (PC-AD) Normal cognition + BM MCI-AD MCI + BM AD dementia (AD-Dem) Dementia + BM Sperling et al., Albert et al., McKhann et al., ADAD 2011
 Molecular. CSF Aβ and tau. PET amyloid imaging. Structural imaging. Hippokampal volumetry. Cortical thickness. Metabolic imaging. FDG-PET. Aβ accumulation markers. CSF Aβ and amyloid imaging. Neurodegeneration markers. CSF tau and structural/metabolic imaging. Pre-clinical AD (PC-AD) Normal cognition + BM. MCI-AD. MCI + BM. AD dementia (AD-Dem) Dementia + BM. Sperling et al., Albert et al., McKhann et al., ADAD")
35
Aβ Leaves the Stage to Tau as AD Progresses from Pre-Clininical to Clinical Stages
BOS-Aβ42 Amiloid PET BOS-Tau FDG-PET MRG NP GYA Jack et al. Lancet Neurol 2010

36
PC-AD = Cerebral Amyloidosis
Phase I Evidence of Aβ accumulation Normal cognition (COG ∅) Phase II Positivity of neurodegeneration (ND) markers Phase III Subjective memory problems (COG +) Phase 0 Aβ ∅, ND ∅, COG ∅ SNAP (suspected non-Alzheimer pathology) Aβ ∅, ND +, COG +/- Unclassified (Phase X) Aβ +, ND -, COG + Aβ ∅, ND ∅, COG + Sperling et al., Alzheimer Dement 2011
 Phase II. Positivity of neurodegeneration (ND) markers. Phase III. Subjective memory problems (COG +) Phase 0. Aβ ∅, ND ∅, COG ∅ SNAP (suspected non-Alzheimer pathology) Aβ ∅, ND +, COG +/- Unclassified (Phase X) Aβ +, ND -, COG + Aβ ∅, ND ∅, COG + Sperling et al., Alzheimer Dement")
37
PC-AD Outcomes Mayo Series
529 HS aged between years MRI, FDG-PET, PIB-PET Phase 0 : 44% Phase I : 16% Phase II : 12% Phase III : 2% SNAP : 23% Phase X : 3% 1-year follow-up of 296 cases: 31 MCI/Dem cases Phase 0 : 5% Phase I : %11% Phase II : 21% Phase III : 43% SNAP : 10 % Evre X : 10% Knopman et al., Neurology 2012

38
PC-AD Outcomes Washington Uni Series
65 years +, 311 HS BOS Aβ, τ; Phase 0 : 41% Phase I : 15% Phase II : 12% Phase III : 4% SNAP : 23% Phase X : 5% CDR 0.5 in 5 years Phase 0 : 2% Phase I : 11% Phase II : 26% Phase III : 56% SNAP : 5 % Evre X : 29% Vos et al., Lancet Neurol 2013

39
Subjective Memory Complaints and Amyloid Deposition
131 HS, 73.5±6 years of age; Boston, ABD (Amariglio et al., Neuropsychologia 2012) Amyloid load as detected by PIB-PET Correlates with subjective memory complaints Does not correlate with objective memory and executive scores.
 Amyloid load as detected by PIB-PET. Correlates with subjective memory complaints. Does not correlate with objective memory and executive scores.")
40
Age-Associated Memory Impairment (AAMI)
AAMI: Poor memory performance of >50 year-old subjects as compared to young (<30) individuals (NIA 1985) Individuals with AAMI cannot acquire the contextual details of the recently learned information (Zacks et al. 2000) Contextual similarity increases the memory difficulty (Henkel et al. 1998) Responding to novelty weakens The ability to discriminate novel and familiar items decreases (Friedman 2000) Eelectrophysiologically the latencies and amplitudes of the novelty P3a responses in ERP recording prolongs and decreases respectfully (Daffner et al. 2005)
 individuals (NIA 1985) Individuals with AAMI cannot acquire the contextual details of the recently learned information (Zacks et al. 2000) Contextual similarity increases the memory difficulty (Henkel et al. 1998) Responding to novelty weakens. The ability to discriminate novel and familiar items decreases (Friedman 2000) Eelectrophysiologically the latencies and amplitudes of the novelty P3a responses in ERP recording prolongs and decreases respectfully (Daffner et al. 2005)")
41
AAMI: Compensation of Novelty Detection Failure by Familiarity
Dentate gyrus Mossy fibres Perforant pathway ECII Multimodal Association Cortices ECIII ECIV-VI CA3 Pattern discrimination vs. Pattern completion Schaffer collaterals Subiculum CA1 Oto-associative fibres Wilson et al. TINS 2006

42
Variation on the Theme of NIA-AA
Alzheimer’s cerebral amyloidosis (Pre-clinical AD) Biomarker positivity with normal cognition Anti-amyloid + other prevention Alzheimer’s neurodegeneration (Clinical AD) Mild (AD-AAMI) Biomarkers + novelty detection failure (familiarity compensation) Anti-amyloid (?) + anti-tau + glutamate modulation Moderate (AD-MCI) Biomarkers + isolated episodic memory impairment Anti-amyloid + anti-tau + Glu modulation ± Cholinergic replacement (AChEIs) Severe (AD-dementia) Biomarkers + multiple cognitive deficits Anti-tau + glutamate modulation ± Cholinergic replacement (AChEIs)
 Biomarker positivity with normal cognition. Anti-amyloid + other prevention. Alzheimer’s neurodegeneration (Clinical AD) Mild (AD-AAMI) Biomarkers + novelty detection failure (familiarity compensation) Anti-amyloid ( ) + anti-tau + glutamate modulation. Moderate (AD-MCI) Biomarkers + isolated episodic memory impairment. Anti-amyloid + anti-tau + Glu modulation ± Cholinergic replacement (AChEIs) Severe (AD-dementia) Biomarkers + multiple cognitive deficits. Anti-tau + glutamate modulation ± Cholinergic replacement (AChEIs)")
43
Possible Benefits of Discriminating Between Alzheimer’s Amyloidosis and Neurodegeneration
CA-ND relationship is probably not a linear sequential inevitability (Lazarczyk et al., BMC Med 2012) Progression from CA to ND is preventable either by natural defence mechanisms and/ or pharmacologically. Analogies with atherosclerosis and MI/stroke, osteoporosis and pathological fracture The treatment of AD-CA and AD-ND will probably be different , if with some overlaps in the future. CA: anti-amyloid + other prevention ND: : anti-amyloid (?early stage) + anti-Tau + established AD treatment
 Progression from CA to ND is preventable either by natural defence mechanisms and/ or pharmacologically. Analogies with atherosclerosis and MI/stroke, osteoporosis and pathological fracture. The treatment of AD-CA and AD-ND will probably be different , if with some overlaps in the future. CA: anti-amyloid + other prevention. ND: : anti-amyloid ( early stage) + anti-Tau + established AD treatment.")
Benzer bir sunumlar

>")

 DİPLOMA EKİ EĞİTİM SEMİNERİ 2011-2013 Dönemi Bologna Sürecinin Türkiye’de.>")










 (Yrd. Doç. Dr. Deniz Dal)>")



>")
