Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
Anabilim Dalı Hematoloji Bilim Dalı Olgu Sunumu 9 Ekim 2014 Perşembe Konuk Ar. Gör. Dr. Esra Ülgen Temel

2
ÇOCUK HEMATOLOJİSİ ARAŞ. GÖR. DR. Esra ÜLGEN TEMEL
Olgu sunumu ÇOCUK HEMATOLOJİSİ ARAŞ. GÖR. DR. Esra ÜLGEN TEMEL

3
Olgu 1 6 yaşında kız hasta iştahsızlık, sık enfeksiyon geçirme, kansızlık şikayetleri ile başvurdu.

4
ÖYKÜ Hasta 15 aylıktan itibaren anemi nedeni ile değişik merkezlerde tetkik edilmiş. Birkaç kez oral demir tedavisi verilmiş. Son olarak, başvurusundan 8-9 ay önce demir tedavisi almış, yanıt olmadığı görülünce tedavi kesilmiş.

5
Öz geçmiş: Perinatal sorun yaşanmamış. Önemli bir hastalık öyküsü yok.
Soy geçmiş: Anne- baba arasında akrabalık yok. 1. gebelik 15 yaşında kız sağlıklı 2. gebelik hastamız. Ailede bilinen hematolojik hastalık öyküsü yok.

6
FİZİK MUAYENE Genel durumu iyi. KTA: 80/dk Ağırlık: 20.5 kg (25-50p)
Boy: 117 cm (50-75p) Cilt: Soluk Kalp: Ritmik, üfürüm yok. Karın: Organomegali yok, traube açık. Solunum Sis. : Normal KBB: Normal Ekstremiteler: Normal Genitoüriner Sis. : Haricen kız
 Cilt: Soluk. Kalp: Ritmik, üfürüm yok. Karın: Organomegali yok, traube açık. Solunum Sis. : Normal. KBB: Normal. Ekstremiteler: Normal. Genitoüriner Sis. : Haricen kız.")
7
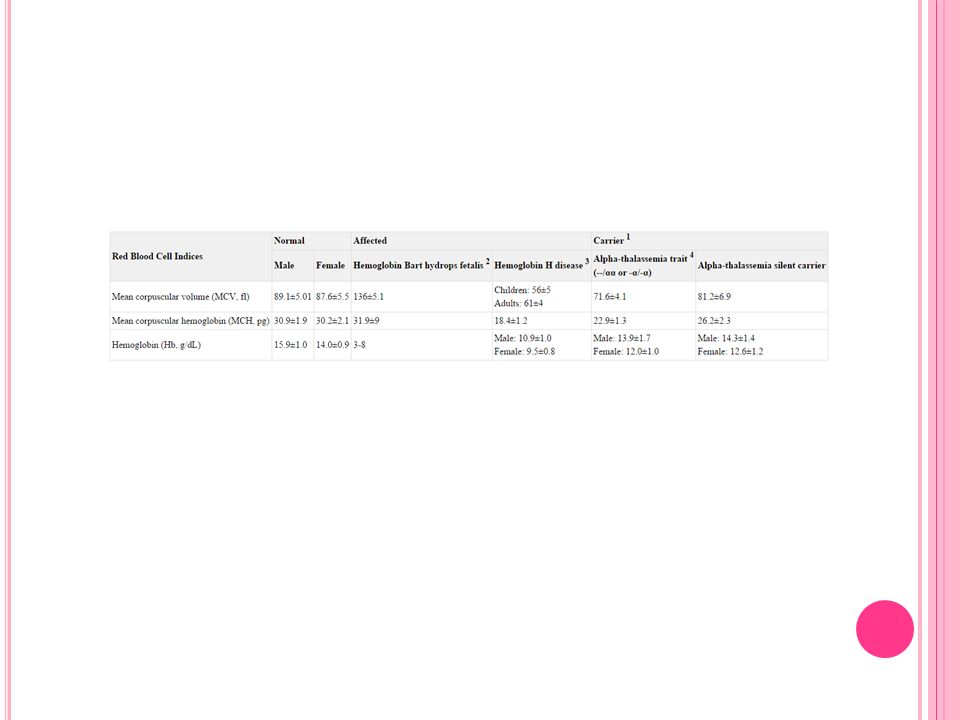
HB 7.3 g/dl 7.7 g/dl 6.3 g/dl 7.4 g/dl RBC 4.49 x10^6/ul 5.3 x10^6/ul 3.78 4.22 MCV 50.9 fL 50.6 fL 58 fL PLT 360000/ul /ul /ul WBC 12300/ul 8890/ul 9100/ul 5800/ul Ferritin Tüm değerlendir melerde normal sınırlarda

8
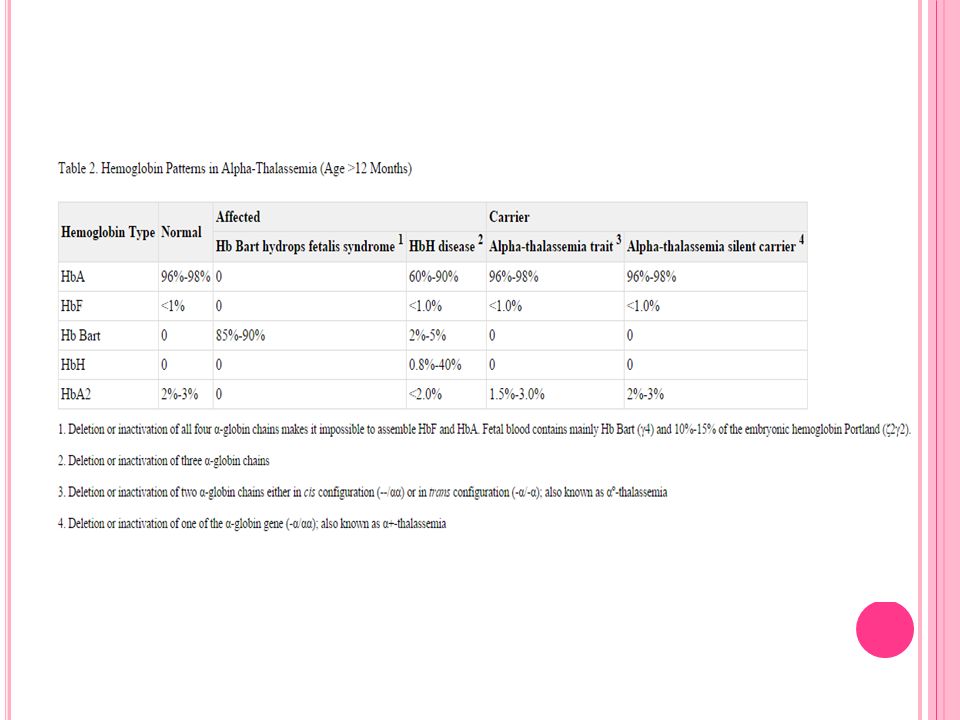
Hemoglobin elektroforezi
Hb A Hb F Hb A2 %1.8 %2 %78.1 %2.1 %0.6 %98.5 %0.5 %1

9
WBC 6680 /uL T. Bil. 1.6 mg/dl HB 7.38 g/dl Ferritin 89 ng/ml PLT /uL LDH 317 U/L MCV 55.8 fL Krea. 0.51 mg/dl RBC 4.15x10^6/uL Retikülosit %1.82

10
Periferik yayma: Hipokrom mikrositer eritrositler. Anizositoz, poikilositoz mevcut. Target cell, göz yaşı hücreleri, tek tük orak hücreler görüldü. %36 PNL %56 lenfosit %2 bant %6 monosit

11
Olgu 2 3 yaşında erkek hasta Azerbaycan da yapılan tetkiklerinde bir kan hastalığı olduğu söylenmiş. Aile kendi isteği ile ileri tetkik ve tedavi amaçlı başvurdu.

12
ÖYKÜ Hastanın yapılan tetkiklerinde Hb düşük bulunuyormuş.
Sık hasta oluyormuş.

13
Öz geçmiş: Miadında, NSVD ile 3200 gr olarak doğmuş
Öz geçmiş: Miadında, NSVD ile 3200 gr olarak doğmuş. YD sarılığı nedeni ile fototerapi almış. Bulunduğu ülkede aşıları yapılmamış. Soy geçmiş: Anne- baba arasında akrabalık yok. 1. gebelik hastamız Ailede bilinen hematolojik hastalık öyküsü yok.

14
FİZİK MUAYENE Genel durumu iyi. KTA: 80/dk Ağırlık: 14 kg (25-50p)
Boy: 95.5 cm (50-75p) Cilt: Soluk Kalp: Ritmik, 1/6 midsistolik üfürüm. Karın: Organomegali yok, traube açık. Solunum Sis. : Normal KBB: Normal Ekstremiteler: Normal Genitoüriner Sis. : Haricen erkek. Testisler skrotumda.
 Cilt: Soluk. Kalp: Ritmik, 1/6 midsistolik üfürüm. Karın: Organomegali yok, traube açık. Solunum Sis. : Normal. KBB: Normal. Ekstremiteler: Normal. Genitoüriner Sis. : Haricen erkek. Testisler skrotumda.")
15
WBC 9970/uL T. Bil. 1.0 mg/dl HB 7.8 g/dl Ferritin 39 ng/ml PLT /ul LDH 232 U/L MCV 55.8 fL Krea. 0.59 mg/dl RBC 4.71 x10^6/ul Retikülosit % 1.7

16
Periferik yayma: Hipokrom mikrositer eritrositler. Anizositoz, poikilositoz mevcut. Target cell, göz yaşı hücreleri mevcut. % 42 PNL % 52 lenfosit % 4 bant % 2 eozinofil

17
ÖN TANILAR

18
TANI Alfa talasemi (Hb H hastalığı )
")
19
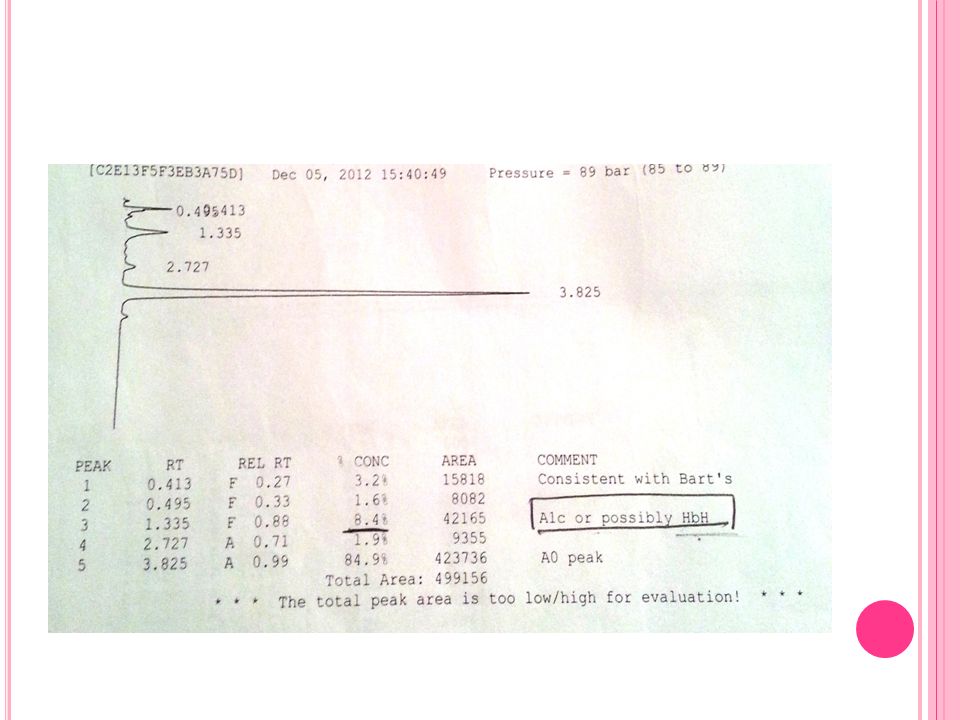
Olgu 1 Hemoglobin elektroforezi Hb A Hb Barts Hb H 05.11.2012 %84 %3.2
%8.4

22
Olgu 2 Hemoglobin elektroforezi Hb A Hb A2 Hb H 11. 09.2014 %83 %0.8
%10.3

24
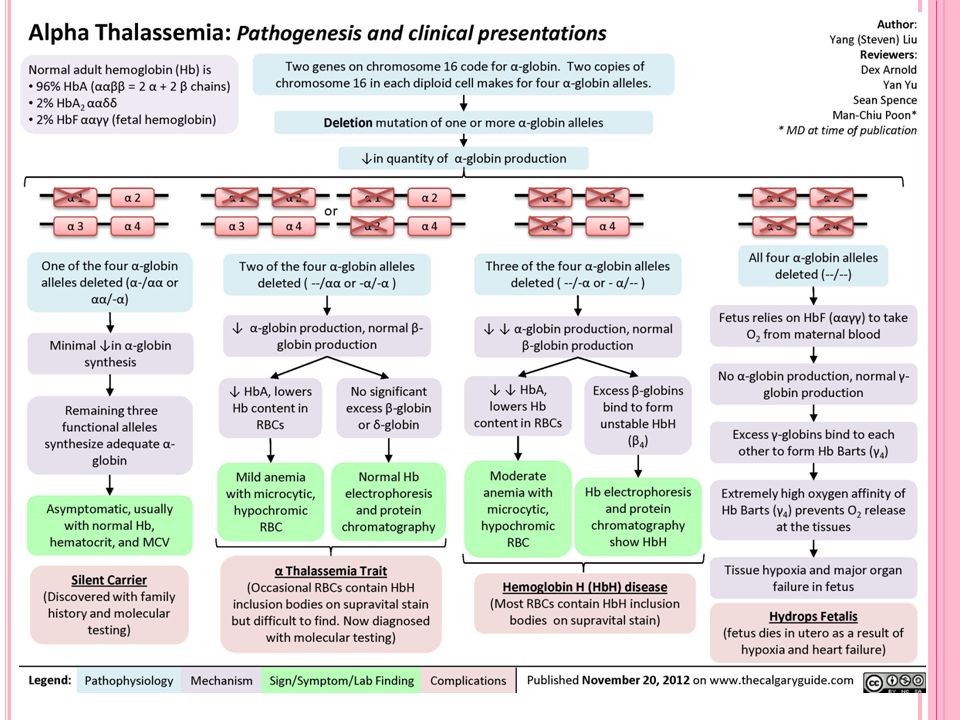
ALFA TALASEMİ

26
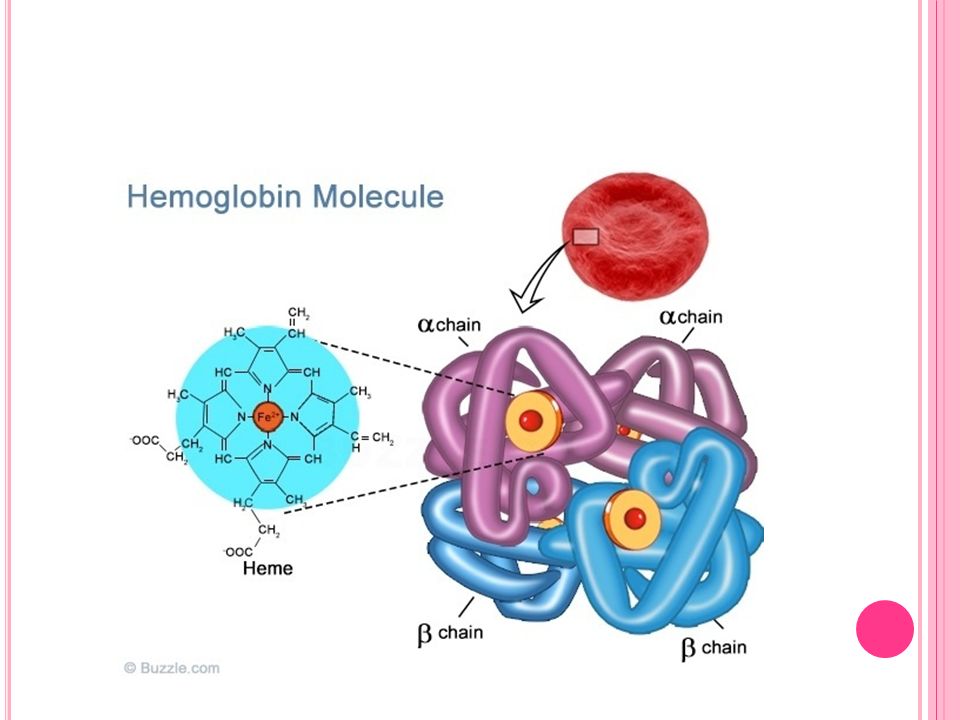
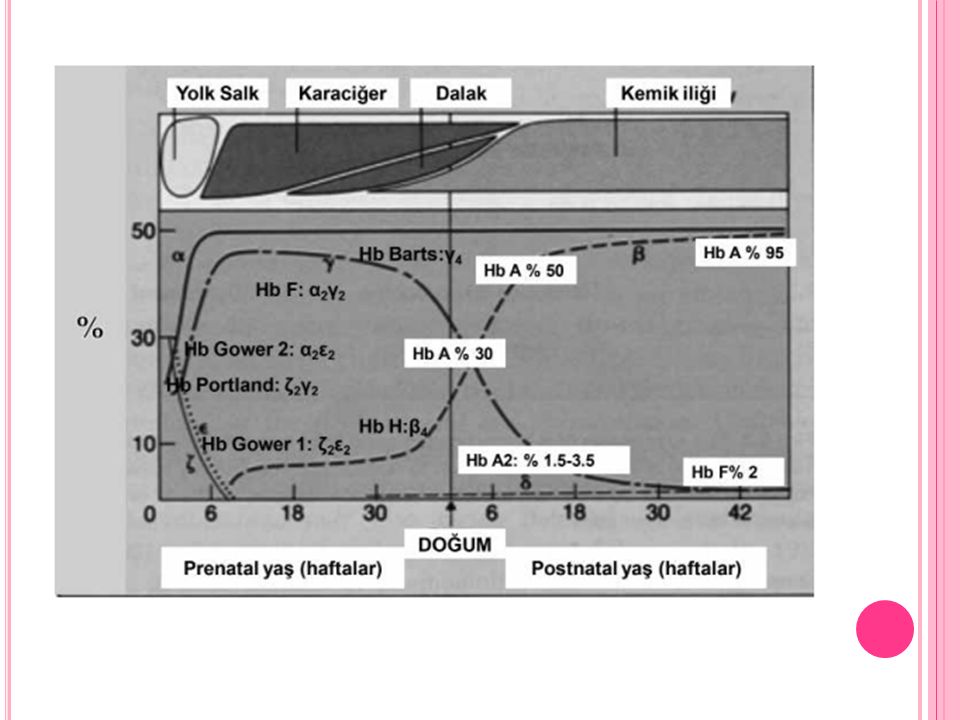
Normal Hemoglobinler 1. HbA1: Yetişkin insan Hb’ninin % kadarını oluşturur, 2α ve 2β zincir içerir. HbA1’in alt grupları vardır. Bunlardan HbA1c hemoglobinin glukoz bağlamış formudur. 2. HbA2: Yetişkin insan Hb’ninin % 2 kadarını oluşturur, 2α ve 2δ zincir içerir

27
Gebeliğin ilk 3 ayında; Gower I Hemoglobin 2 zeta 2 epsilon Gower II Hemoglobin 2 alfa 2 epsilon Portland Hemoglobin 2 gamma 2 zeta Son 6 ayda: HbF (fetal Hb): 2α ve 2γ zincir içerir. Yetişkin insanlarda da az miktarda bulunur
: 2α ve 2γ zincir içerir. Yetişkin insanlarda da az miktarda bulunur.")
29
Anormal Hemoglobinler
Genetik hemoglobinopatilerdir Normal ve anormal hemoglobinler arasındaki fark amino asit dizilişlerinin farklılığı ve buradan kaynaklanan elektroforetik göç hızı farklılığıdır Anormal Hb tipleri en çok HbA1‘de görülür

30
Anormal hemoglobinler 3 temel tipte olabilir
A) Peptid zincirlerindeki amino asit diziliş farklılığı Yalnız α zincirde değişlik var (Toronto, Honolulu, Osaka….) Yalnız β zincirde değişlik var (HbS, orak hücre anemisi: 6. aa olan glutamik asit yerine valin gelmiştir; HbC; HbI) Hem grubu azalması var (mutasyon nedeniyle Fe2+ yerine Fe3+ bağlanır, metHb oluşur, oksijen bağlamaz) B) Peptid zincirlerinin 4’ünün de aynı olması (HbH: 4β; HbBart:4γ) C) Zincirlerden birinin eksik sentezlenmesi (talasemiler)
 Peptid zincirlerindeki amino asit diziliş farklılığı. Yalnız α zincirde değişlik var (Toronto, Honolulu, Osaka….) Yalnız β zincirde değişlik var (HbS, orak hücre anemisi: 6. aa olan glutamik asit yerine valin gelmiştir; HbC; HbI) Hem grubu azalması var (mutasyon nedeniyle Fe2+ yerine Fe3+ bağlanır, metHb oluşur, oksijen bağlamaz) B) Peptid zincirlerinin 4’ünün de aynı olması (HbH: 4β; HbBart:4γ) C) Zincirlerden birinin eksik sentezlenmesi (talasemiler)")
32
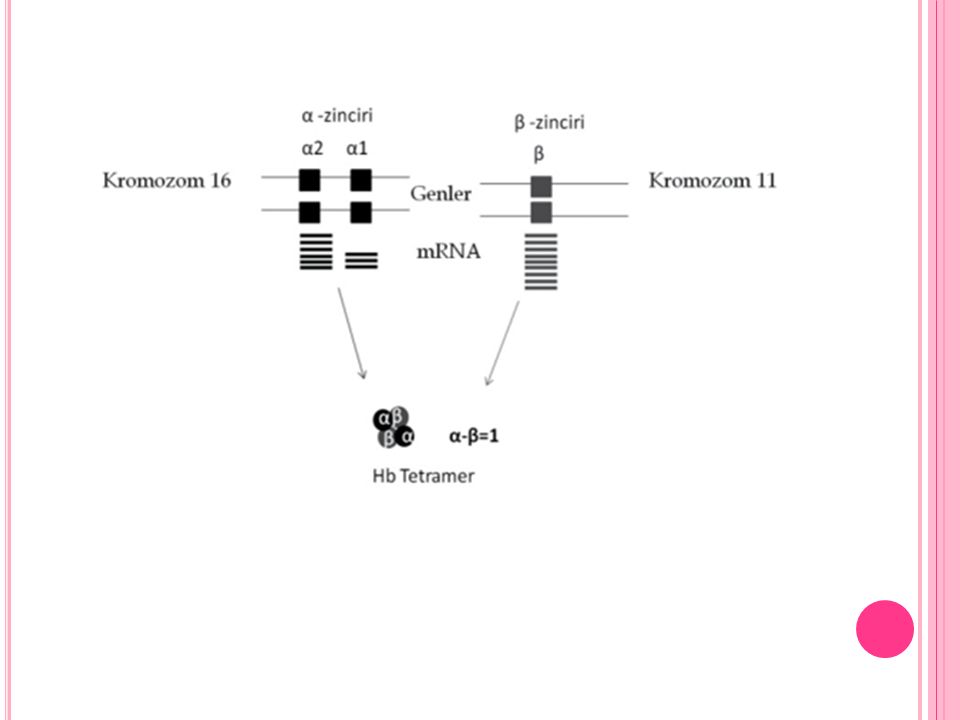
Bugüne kadar 500’den fazla α globin geni mutasyonu bildirilmiştir
Bugüne kadar 500’den fazla α globin geni mutasyonu bildirilmiştir. Bu mutasyonlar 3 ana grupta toplanır: 1. Delesyonlar: Alfa talasemide en sık rastlanılan hastalık sebebidir. Delesyonların genişliği önemlidir ve klinik fenotipi etkilemektedir. 2. Nokta mutasyonları: Beta talasemide sıktır, alfa’da nadirdir. 3. Düzenleyici elementlerde nadir delesyonlar.

35
Kalitatif ve kantitatif hemoglobin analizi (selluloz asetat elektroforezi, weak-cation HPLC ve izoelektrik odaklama ve sitrat agar elektroforezi gibi tamamlayıcı teknikler.) ile hemoglobin tipleri gösterilebilir.
 ile hemoglobin tipleri gösterilebilir.")
37
Hb BART’s SENDROMU Etkilenen fetus HbF ve A yapmak için herhangi bir α-globin zinciri üretemez. Fetal kan başlıca Hb Bart’s (γ4) içerir. Değişken miktarda bulunan Hb Portland O2 taşıyarak bebekleri canlı tutar. Ağır intrauterin anemi kalp yetmezliğine yol açar.
 içerir. Değişken miktarda bulunan Hb Portland O2 taşıyarak bebekleri canlı tutar. Ağır intrauterin anemi kalp yetmezliğine yol açar.")
38
Belirgin hepatosplenomegali, yaygın ödem, asit, iskelet ve kardiyovasküler deformiteler gelişir. Plasenta genişler. Doğumdan sonra ağır anemiye bağlı gelişen şok tedavisinde kan transfüzyonu, yaşam destek tedavisi gerekir yoksa kısa sürede bebek kaybedilir.

39
Gebeliğin erken döneminde tanı konulursa intrauterin transfüzyonlarla gebelik takip edilip erken doğum yaptırılır. Hasta doğum sonrası düzenli transfüzyon ve demir şelasyonu ile izlenir. Gebelik sırasında maternal komplikasyonlar pre eklampsi, polihidramnios, kanama, anemi ve sepsis gelişebilir.

40
Literatürde bildirilen yaşayan Hb Bart’s hidrops fetalis vakaları çok azdır. Bu vakalarda birlikte iskelet ve ürogenital sistem anomalileri, ağır nörolojik bozukluklar bildirilmiştir.

41
Hemoglobin H (Hb H) Hastaliği
Orta-ağır hemolitik anemiye yol açan klinik olarak çok değişken bir grup hastalıktır. Bazı hastalarda hiç transfüzyon gereksinimi olmazken (Transfüzyona bağımlı olmayan talasemi; NTDT), bazılarında aralıklı hatta düzenli transfüzyon gerekebilir (Transfüzyona bağımlı talasemi).
, bazılarında aralıklı hatta düzenli transfüzyon gerekebilir (Transfüzyona bağımlı talasemi).")
42
Hb H hastalığının en sık nedeni 3 alfa globin genindeki delesyondur (-α/--). Diğerleri ise, α1 veya α2 geninde 2 gen delesyonu ve bir nokta mutasyonu (αTα/--) (ααT/--) veya özellikle α2 geninde ağır nokta mutasyonlarıdır (αTα/ αTα) (Nondelesyonel Hb H).
 (ααT/--) veya özellikle α2 geninde ağır nokta mutasyonlarıdır (αTα/ αTα) (Nondelesyonel Hb H)..")
44
Hb H hastalığının klinik şiddeti esas olarak alfa genindeki mutasyonlara bağlı olsa da beta talasemi ve unstabil beta globin varyantları (anormal hemoglobin) birlikteliği de klinik tablonun şekillenmesinde etkilidir. Aynı alfa talasemi mutasyonuna sahip olan hastaların bazıları hafif, bazıları orta hatta bazıları ağır derecede anemiye sahip iken beta talasemi mutasyonu birlikteliğinde, alfa ve beta zincirleri arasındaki dengesizlik azaldığı için klinik hafifleyebilmektedir.

45
Alfa gen triplikasyonu ve heterozigot β-talasemi mutasyonu birarada olduğunda β-talasemi taşıyıcılığından daha ağır bir anemi ve Hb F yüksekliği (%2-15 arasında) gösteren talasemi intermedia tablosu görülür. Çocukta Talasemi İntermedia kliniği olup aile çalışmasında anne ve babadan sadece birinde β- talasemi taşıyıcılığı olması bu olasılığı düşündürmelidir.

46
Fizyopatoloji Alfa-talasemide β-zinciri göreceli olarak α- zincirinden daha fazla yapılır. Fazla olan beta zincirleri tetramer yapar, hem ile bağlanır ve durağan olmayan bir hemoglobin olan Hb H meydana gelir.

47
Dayanıksız bir hemoglobin olan hemoglobin H’nin eritrositler içinde çökmesine bağlı olarak hemolitik anemi gözlenir. Periferik kanda retikülosit boyası ile bu presipitasyonlar gösterilebilir (Hb H inklüzyon cisimcikleri).
.")
49
Kemik iliğinde ise eritroid ana hücrelerin olgunlaşma süreci normal olup diseritropoez yoktur, dolayısı ile genellikle periferik yaymada normoblast görülmez. Retikülosit sayısı ise anemiyle orantılı olacak şekilde yüksek olabilir

50
İzlem Orta derecede anemiyle seyreden Hb H hastalarında artmış kemik iliği aktivitesi ve artmış oksidatif stresi düzenlemek için folik asit (5 mg/gün), D vitamini ve E vitamini, kalsiyum ve çinko desteği verilmelidir. Ağır nondelesyonel Hb H hastalarında (Hb PNP, Hb Adana, vb.) ağır anemi olabilir. Altı yaşın altındaki ağır anemi vakalarında düzenli transfüzyon ve şelasyon tedavisi uygulanmalı, talasemi major gibi takip ve tedavi edilmelidir
, D vitamini ve E vitamini, kalsiyum ve çinko desteği verilmelidir. Ağır nondelesyonel Hb H hastalarında (Hb PNP, Hb Adana, vb.) ağır anemi olabilir. Altı yaşın altındaki ağır anemi vakalarında düzenli transfüzyon ve şelasyon tedavisi uygulanmalı, talasemi major gibi takip ve tedavi edilmelidir.")
51
Splenektomi transfüzyon ihtiyacını azaltabilir ancak ileriki yaşlarda tromboz ve vaskülopati riskini arttırdığından yalnızca seçilmiş, gerekli hastalarda uygulanmalıdır. Splenektomi öncesi H. influenza, pnömokok ve meningokok aşıları yapılmalı ve her 5 yılda bir düzenli olarak tekrarlanmalıdır. Ayrıca bu hastalara 3-5 yıl süreyle penisilin profilaksisi yapılmalıdır (penisilin V, 20 kilonun üzerinde 250 mg/gün, günde 2 kez olacak şekilde). Splenektomi sonrası trombositoz gelişeceğinden tromboz profilaksisi için aspirin (80 mg/gün) verilmelidir
. Splenektomi sonrası trombositoz gelişeceğinden tromboz profilaksisi için aspirin (80 mg/gün) verilmelidir.")
52
Düzenli transfüzyon almayan Hb H hastalarında enfeksiyon, ateş, oksidatif stres, gebelik durumlarında gelişen (akut hemolitik kriz nedeniyle) transfüzyon ihtiyacı olabilir. Bu sırada ciddi sarılık, hemoglobinemi, hemoglobinüri olduğunda böbrek hasarı ve akut böbrek yetmezliği gelişebilir. Bu durumda acil tedavi gerekir, iv sıvı (idrar alkali olacak şekilde) verilmeli, enfeksiyon varsa ampirik antibiyotik başlanmalı, transfüzyon yapılmalıdır.
 verilmeli, enfeksiyon varsa ampirik antibiyotik başlanmalı, transfüzyon yapılmalıdır.")
53
İLİŞKİLİ DİĞER SENDROMLAR
ATR 16 Kromozom 16 üzerinde α talasemi/mental retardasyon sendromu (OMIM:141750)’dur. Alfa globin genleri ile birlikte onun etrafındaki diğer genlerin de etkilendiği çok büyük delesyonlar zihinsel yetersizliğin de eşlik ettiği gelişimsel anomalilerle ilişkilendirilir.
’dur. Alfa globin genleri ile birlikte onun etrafındaki diğer genlerin de etkilendiği çok büyük delesyonlar zihinsel yetersizliğin de eşlik ettiği gelişimsel anomalilerle ilişkilendirilir.")
54
ATR-X Alfa talasemi ile ilişkili X’e bağlı mental retardasyon sendromu (OMIM:301040)’dur. AT-MDS α talasemiye neden olan ATRX genindeki mutasyonlarla oluşan bir sendromdur. (OMIM:300448). Ender görülen bu sendroma MDS eşlik eder. Özellikle MDS’li yaşlı erkeklerde görülür
. Ender görülen bu sendroma MDS eşlik eder. Özellikle MDS’li yaşlı erkeklerde görülür.")
55
TEŞEKKÜRLER

Benzer bir sunumlar















>")







