Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
Anabilim Dalı Yenidoğan Bilim Dalı Olgu Sunumu 9 Nisan 2013

2
Sabah ToplantIsI Yenİdoğan Vaka Sunumu
Dr. Özge YENDUR Uzm.Dr. Evrim ÇELİKER YAPICI Prof.Dr.Ayşe Engin ARISOY

3
11 saatlİk, erkek bebek, 32 yaşında ,Gestasyonel diabetli anneden, G3P1(ikiz)Y1D0K1, 36+4 Gestasyon haftalık, 3290 gr , dış merkezde, sezeryan ile, Doğar doğmaz ağlamamış , KTA:40/dk. PPV uygulanmış, sonrasında nazal CPAP alınmış. 2. saatte Solunumu düzelmesi üzerine hood içi O2 de izlenmeye başlanmış. Saatler içerisinde solunumunun giderek kötüleşmesi ve hastanın fenotipik patolojik bulguları nedeniyle ileri tetkik ve tedavi amacıyla tarafımıza yönlendirildi.

4
Prenatal: Aylık takipli, fetal USG ler N
Soygeçmiş: Anne 32 y, ss Baba 30 y, ss Akrabalık yok G1: Küretaj, 2. ayda, G2: Hastamız, ikiz eşi birinci G3: ikiz eşi, erkek

5
Fİzİk Muayene BÇ: 35.8 cm (>90p) Boy: 51 cm (90 p)
SS:62/dk , hood içi O2 de spontan soluyor KTA:122/dk Vücut ısısı: 36.1 C TA:Sağ Kol: 96/41 mmHg Sağ Bacak: 95/53 mmHg Sol Kol:85/55 mmHg Sol Bacak: 81/54 mmHg BÇ: 35.8 cm (>90p) Boy: 51 cm (90 p) DA: 3290 gr(75-90p)
 Boy: 51 cm (90 p) DA: 3290 gr(75-90p)")
6
Fİzİk Muayene-Patolojİk Bulgular
Genel durum; Orta , fasiyal dismorfizm.. Cilt rengi doğal, ödem ikter, siyanoz yok. BAŞ BOYUN: Koronal sutur kapalı, yüksek belirgin alın, kraniosinoztosiz

7
Fİzİk Muayene-Patolojİk Bulgular
GÖZLER: Red refleksi +/+ Hipertelorizm, propitozis, Antimongoloid gözler KBB: burun basık , septal deviasyon ,sol burun pasajı kapalı, sağ burun pasajı açık Düşük kulak

8
Yüksek arklı dar damak bifid uvula dentin kistler

9
Fİzİk Muayene-Patolojİk Bulgular
Solunum Sistemi: Retraksiyon+, ral + , toraks deformitesi yok. Kardiyovasküler Bakı: 1/6 sistolik üfürüm, KTA 4. İKA solda . GİS ,GÜS: özellik yok. Ekstremiteler:Bilateral üst ve alt ekstremitelerde sindaktili. NMS: Yenidoğan refleksleri zayıf. Emme refleksi yok.

10
Ekstremiteler: Bilateral üst ve alt ekstremitelerde sindaktili.

11
Laboratuvar: Hemogram, Biyokimya doğal sınırlarda.
Kanama profili: Normal Tiroid fonksiyon testleri normal.

12
Akciğer grafisi normal
EKO:ince duktus açıklığı TFUSG: Bilateral koroid plexusta kistik lezyon. Kanama sekeli olabilir. Sol lateral ventrikül oksipital alanda hemoraji ile uyumlu hiperekojen lezyon Batın USG: Karaciğerde segment 4A-4B lokalizasyonunda portovenöz şant . Mesane glob vezikal izlendi. Üst sınırda mesane duvar kalınlığı.

13
ÖN TANINIZ NEDİR? İkiz bebek, akrabalık yok , Dismorfik yüz görünümü
Yüksek belirgin alın, kraniosinoztosiz Basık burun , septal deviasyon ,koanal atrezi Düşük kulak Yüksek arklı dar damak , bifid uvula , dentin kistler Bilateral üst ve alt ekstremitelerde sindaktili… ÖN TANINIZ NEDİR?

14
APERT SENDROMU

15
APERT SENDROMU İlk olarak 1906 yılında Eugene Apert tarafından tanımlanmıştır. Nadir görülen, kongenital bir malformasyon olup, kutanöz ve kemik sindaktili, midfasial hipoplazi ve kraniosinositoz triadı ile karakterizedir. 10. kromozomda yer alan FGFR2 geninin mutasyonu sonucu gelişir.

16
Literatürde bildirilen vakalar genellikle sporadiktir ve yeni mutasyon nedeniyle oluşmuştur. Otozomal dominant geçiş de bildirilmiştir. Hastalığın ileri baba yaşı ile ilişkili olduğu düşünülmektedir. Sıklığı yaklaşık 1 milyon canlı doğumda arasında değişmektedir.

17
Apert sendromunun kesin ayırıcı tanısı DNA analizi ile yapılabilir.
FGFR2 genindeki 252 ve aminoasitlerdeki değişmeler Apert sendromuna yol açmaktadır.

18
Apert Sendromunda tanımlanan bulgular:

20
Yenidoğanlarda koronal sinositoz nedeniyle kranial fossanın ön-arka boyu kısadır ve sfenoid kemiğin büyük kanatlarının öne doğru yer değiştirmesi nedeni ile alın dik, geniş ve düz, temporal bölgeler çıkıntılı ve oksiput düzdür. Maksiller kemik üç düzlemde gelişemez ve maksiller yükseklik, nazal kavite genişliği ve nazofarengeal yükseklik azalır. Bu yapı orofarengeal ve nazofarengeal boşluğun gelişimini ciddi derecede engeller. Buna bağlı respiratuar fonksiyonlar bozulur, uyku apnesi ve ani ölüm gelişebilir.

21
Simetrik sindaktili gözlenir ve en sık iki, üç ve dördüncü parmaklar arasındadır, bir ve beşinci parmaklar serbesttir. İkinci sıklıkta baş parmak hariç diğer parmaklarda füzyon olur.

22
Ayırıcı tanıda Klippel-Feil sendromu ve kraniosinositozla birlikte görülen yaygın genetik bozukluklar bulunmaktadır. Klippel-Feil sendromunun klasik klinik üçlemesi kısa boyun, düşük ense çizgisi, boyun hareketlerinde kısıtlılıktır. Kraniosinositozla birlikte görülen genetik bozukluklar; Crouzon, Carpenter, Chotzen ve Pfeifer Sendromudur.

23
Crouzon sendromu: Özellikle koronal sütürlerin bilateral ve sagittal sütürün erken kapanması sonucu akrosefali, brakisefali, belirgin ekzoftalmus, ptozis, hipertelorism, gaga şeklinde burun, kulak ve damak deformiteleri bulunur.

24
Carpenter Sendromu: Akrosefali, fasial paralizi ve özel bir yüz görünümü vardır.

25
Pfeiffer Sendromu: otozomal dominant genetik defekt FGFR1 ve FGFR2 mutasyonları ile birlikte görülür. Farklı derecelerde kraniosinositoz ve orta hatta hipoplazi görülür. Geniş baş parmak ve anormal geniş ayak başparmağı ve parmaklarda sindaktili eşlik eder.

26
APERT CROUZON CARPENTER PFEIFFER

27
prognoz Kraniosinositoz operasyonu olan hastalarda normal beyin gelişimi şansı olabilir. Ancak cerrahi müdahaleye rağmen, ek beyin anomalileri nedeniyle beyin gelişimi kötü olabilir. Ailenin çabası ve özel eğitim ile normal zekaya ulaşan hastalar bildirilmiştir. Genellikle davranışsal ve duygusal problemler gözlenir. Hayat beklentisi değişken olmakla birlikte, çocukluk dönemini geçiren hastalarda ciddi kalp probleminin olmaması halinde normal yaşam süresi beklenir.

28
Uzun sürelİ İzlem Plastik ve rekonstruktif cerrahi Beyin cerrahisi
Nöroloji Psikiyatri

29
Frontoorbital düzeltme ve kraniyal yapıyı tekrar oluşturmak için cerrahi girişimin 6- 9 ay arasında yapılması önerilmektedir. Çünkü ilk 6 aylık dönemde kemik yapılar frajildir ve şekil veirlmesi güçtür. 9. aydan sonra ise cerrahi işlemden sonra oluşan kemik defektler ossifikasyon gösteremez ve kemik greftler gerekeceği için tedavi zorlaşır.

30
3-5 yaşlar ve adolesan dönemlerde düzeltici operasyonlar..
Sinostoz için rekonstrüksiyon 6 yaş sonrası önerilir.

31
Çoğu mental retarde çok az bir kısmı normal IQ ya sahiptir.
Mental durumları cerrahi tedavi , eşlik eden beyin anomalileri , aile ve çevresel etkenlerden etkilenir. Nörolojik gelişim ve özel eğitim gerektirir.

33
Temel olarak prenatal tanı USG ile kraniostoz ve sindaktilinin gösterilmesi ile düşünülür.
Bulguların görüldüğü gebelik yaşı 16 dan 32. gestasyon haftaya kadar uzanır. Aile öyküsü olan hastalarda bu bulguların USG ile gösterilmesi tanıda yeterlidir. Ancak sporadik olgularda moleküler genetik çalışma ile tanı kesinleştirilir.

34
İzlem Hastaya geldiğinde 70 cc /kg/g den total mayi başlandı. Nazal CPAPta izleme alındı. Ampisilin-Gentamisin başlandı. Saaatler içerisinde solunum sıkıntısı artan hasta entübe edildi. 3. günde pnömotoraks gelişimi nedeniyle Çocuk cerrahisi bölümü tarafından toraks tüpü takıldı. HFO modda izlendi. Ng sonda sağ burundan ilerletilemeyen hasta ek morfolojik görüntüleri nedeniyle KBB bölümü ile konsülte edildi.

35
İzlem Plastik cerrahi bölümü ile sindaktili onarımı operasyonu için görüşüldü. Beyin cerrahisi bölümüne kraniosinostoz için kranioplasti açısından konsülte edildi. BT istendi. Beyin parankim hasarı ve gelişimsel açıdan prognozunu değerlendirmek amacıyla kranial MR planlandı.

36
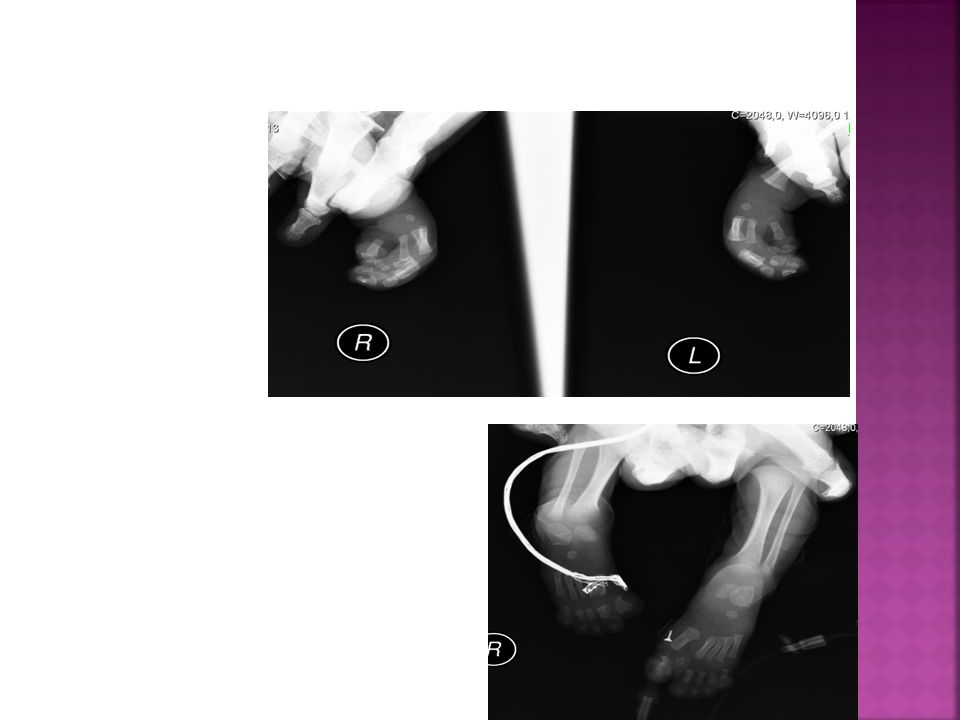
Kafa ve Ekstremite grafileri:

38
İzlem Endotrakeal tüpünden her beslenme sonrasında mama gelişi gözlenmesi nedeniyle antireflü tedavi başlandı. Baş dik pozisyonda tutuldu. Ancak reflüsünün devam etmesi nedeniyle Çocuk cerrahisi bölümü ile görüşülerek radyoopak madde eşliğinde gastrografi çekildi. Özefagusun orta bölümüne kadar uzanan reflü gözlendi.

39
İzlem Özefagusun orta bölümüne kadar uzanan reflü gözlendi.

40
İzlem H tipi fistül açısından skopi altında grafi ya da bronkoskopi ile değerlendirilmesi planlandı. Beslenmenin 24x5 cc kontinü beslenme ile dahi reflünün devamı üzerine oral beslenmesi kesilerek düodenal tüp takılması ya da gereğinde gastrostomi açılması planlandı. Takiplerinde orofarengiyal tüp ile spontan solunması gözlendi ancak tolere edemediği görülünce tekrar entübe edilerek devam edildi.

Benzer bir sunumlar