Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Nöroloji Bilim Dalı Olgu Sunumu 17 Mart 2016 Perşembe Yandal Ar. Gör. Uzm. Dr. Ayfer Sakarya Güneş

2
17.03.2016 Olgu Sunumu Kocaeli Üniversitesi Tıp Fakültesi Çocuk Nöroloji Bilim Dalı Dr.Ayfer Sakarya Güneş

3
1 yaş, Kız bebek Şikayet: Oturamama

4
Özgeçmiş Annenin ilk gebeliği, gebelik sorunsuz geçmiş 38 GH’da, C/S(kanama olması nedeni ile), tartı 3.160 g, boy 50 cm, BÇ 34 cm doğmuş Asfiksi öyküsü yok, 5 gün yd sepsisi nedeni ile küvöz bakımı almış, sarılık öyküsü yok Yakınmaları dışında başka hastalık öyküsü yok Soygeçmiş Anne 28 yaşında, sağlıklı, üniversite mezunu Baba 32 yaşında sağlıklı, lise mezunu 1.Çocuk: Hastamız Anne-baba arasında akrabalık var. (2. derece )
.")
5
Fizik Muayene: Kilo:10kg (50-75p), Boy:73 cm (50p) BÇ: 44,5 cm ( 3-10p) Genel durum: iyi Cilt: Turgor-tonus doğal. Baş-boyun: Saç ve saçlı deri doğal. Kafa yapısı simetrik. Boyunda kitle ve LAP yok. Gözler: Işık refleksi bilateral mevcut. Pupiller izokorik. Konjonktivalar ve skleralar doğal. Heliotrop raş yok. Kulak-Burun-Boğaz: Bilateral kulak zarları doğal. Burun tıkanıklığı,akıntısı yok. Kardiyovasküler: S1-S2 doğal. Üfürüm yok.S3 yok. AFN her iki alt ekstremitede alınıyor. Kalp tepe atımı 5. interkostal aralıkta. Solunum sistemi: Her iki hemitoraks solunuma eşit katılıyor. Ral, ronküs, ekspiryum uzunluğu yok.

6
Gastrointestinal sistem: Batın rahat. Hassasiyet yok.Defans,rebound yok. Hepatomegali, splenomegali yok. Barsak sesleri doğal. Genitoüriner sistem: Haricen kız anomali yok. Nöromusküler sistem: Bilinç açık, koopere –oryante. Kranial sinir muayenesi doğal. Patolojik refleks yok. Hamstringler hafif gergin. Klonus yok. Ekstremiteler: Kas kitlesi doğal, deformite yok. Başını dik tutabiliyor, destekli oturuyor, desteksiz 2-3 sn oturabiliyor, heceleme ve kelimesi var.

7
Laboratuvar: tam kan sayımı rutin biyokimya tam idrar tetkiki; normal doğumsal metabolik hastalık tarama testleri; özellik yok

8
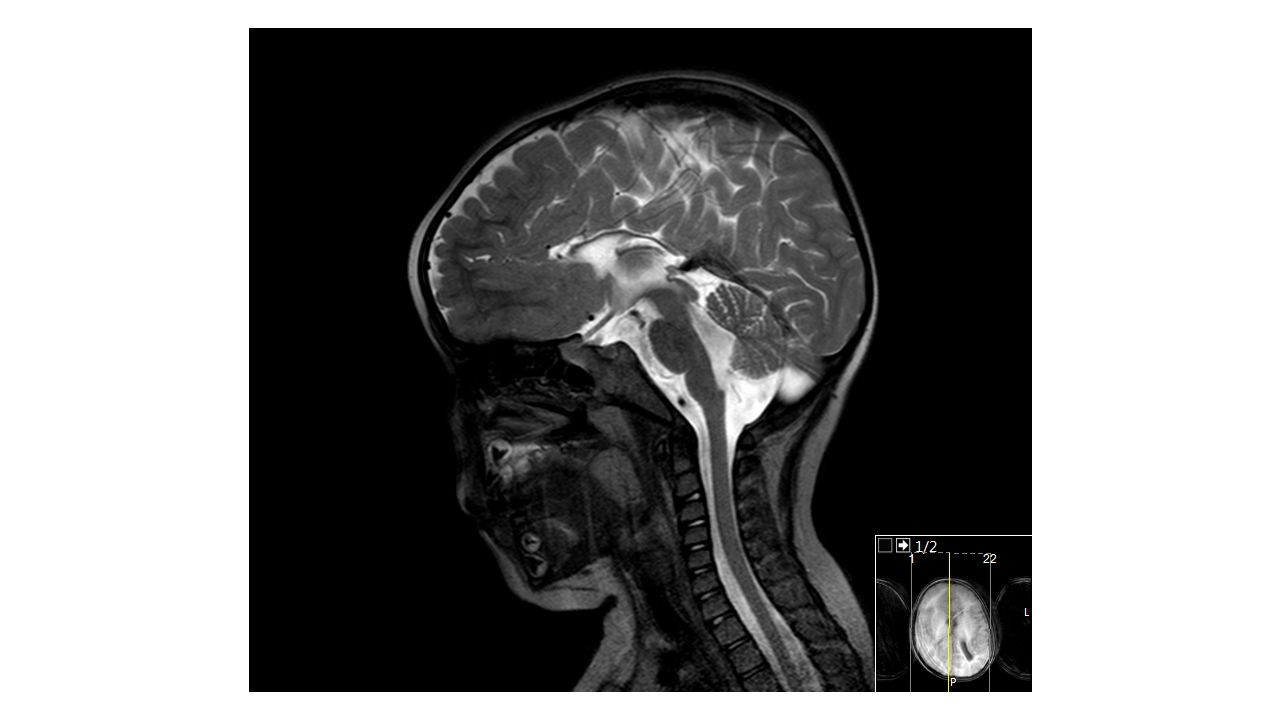
Kranyal MR:

10
Hastanın izleminde; nöbetleri ortaya çıktı, nöbetleri VPA ile kontrol altına alındı, psikomotor geriliği belirgin hale geldi, fizik muayenede, alt ekstremitelerde spastisite, DTR’leri artmış-yayılıyor ve klonus +/+ saptandı.

11
Patolojik bulgular -Oturamama -Yd sepsis nedeni ile 5 gün küvöz bakımı -Hamstringler hafif gergin -Kranyal MR’da ventrikül konturları düzensiz, bilatreal arka horn komşuluğunda T2 sinyal artışı, korpus kallosum hipoplazik

12
Ön Tanı?

13
Ailenin ikinci gebelikten, term doğan bebekleri de 8 aylıkken oturamama yakınmasıyla getirildi. perinatal öyküsü, muayenesi ve kranyal MR görüntüleme bulguları ablasına benzerdi, 16 aylıkken West sendromu tanısı ile 1 kür ACTH tedavisi aldı, nöbetleri VPA ile kontrol altına alındı. Anne ve babanın fizik muayene bulguları normaldi.

14
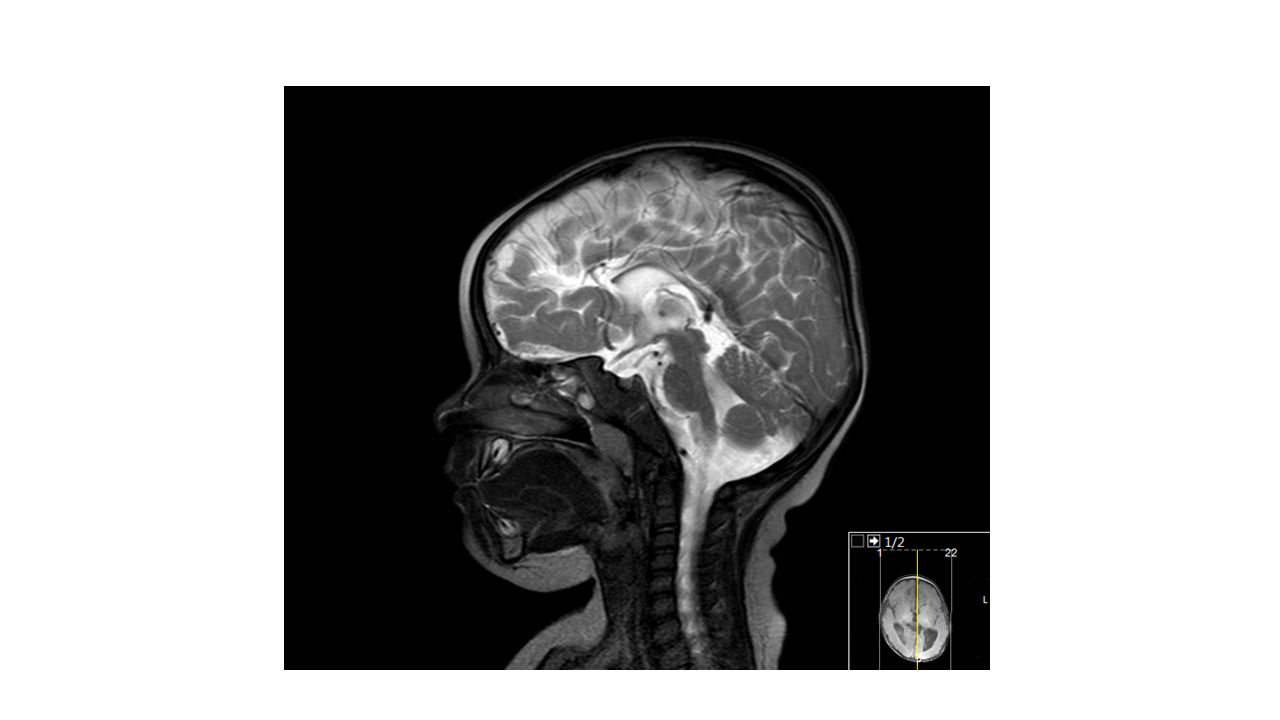
Kranyal MR:

16
Ön tanı?

17
Beyin ve spinal kord ile ilgili yapısal sorunlar - PVL, tethered kord Lökodistrofi - Krabbe Hastalığı, mitokondriyel hastalık Dejeneratif hastalık - HSP

18
Her iki kardeşte de perinatal asfiksi ve/veya prematürite öyküsü olmadığı halde spastik paraparezi lehine klinik bulguları ve periventriküler lökomalaziyle uyumlu kranyal MR bulguları vardı. Spinal MR normal saptandı. Diğer nedenler de ekarte edildikten sonra OR geçişli herediter spastik paraparezi açısından genetik inceleme planlandı.

19
Tüm ekzon analizinde her iki kardeşte EPG5 geninde mutasyon saptandı.

20
Tedavi: Özel eğitim ve fizik tedavi desteği Antiepileptik tedavi

21
Herediter Spastik Paraparezi Herediter spastik paraparezi (HSP), alt ekstremitelerde giderek artış gösteren spastisite, güçsüzlük ve yürüme bozukluğuyla karakterize nörodejeneratif bir hastalıktır. Lateral kortikospinal yollar ve fasikulus grasilis liflerini tutan akson dejerasyonu, Dejenerasyon, bu aksonların distal sonlanım yerlerinde daha ağırdır. - kortikospinal yol (motor); torasik spinal kord - fasikulus grasilis lifleri (duyusal); sevikomedüller bölde
; torasik spinal kord - fasikulus grasilis lifleri (duyusal); sevikomedüller bölde.")
22
Klinik; İzole HSP; sadece spastik parapareziyle seyreder Komplike HSP; progresif olmayan entelektüel gerilik, demans, periferik nöropati, ataksi, ekstrapramidal bulgular, katarakt, optik atrofi gibi farklı nörolojik sorunlar eşlik edebilir

23
Semptomların başlama yaşı infant döneminden erişkin yaşlara kadar değişebilmektedir. Yürüme güçlüğü çok hafif olup fark edilmeyebileceği gibi tekerlikli sandalye gerektirecek kadar ağır spastik parapleji olabilir. Alt ekstemite spastisitesi ve eşlik eden tendon kontraktürleri farklı derecelerde(hafif/şiddetli) de olsa her zaman vardır. - özellikle etkilenen kaslar; hamstring, kuadriseps, gastroknemius-soleus ve adduktor kas grupları Güçsüzlük her zaman eşlik etmez, -ilk etkilenen kaslar; iliopsoas, hamstring ve tibialis anterior
 de olsa her zaman vardır. - özellikle etkilenen kaslar; hamstring, kuadriseps, gastroknemius-soleus ve adduktor kas grupları Güçsüzlük her zaman eşlik etmez, -ilk etkilenen kaslar; iliopsoas, hamstring ve tibialis anterior.")
24
Semptomlar asimetrik olarak başlayabilir ve birkaç yıl içinde her iki alt ekstremite benzer duruma gelir. Mesane fonksiyon bozuklukları sık ve genellikle erken semptom verir. Tipik olarak urgency ile başvuru olur; daha az sıklıkla üriner retansyon da görülebilir. Dış sfinkter bozukluğuna bağlı defekasyon ile ilgili sorunlar da ortaya çıkabilir.

25
Genellikle otozomal dominant geçiş gösterir, ancak son yıllarda otozomal resesif ve X'e bağlı kalıtılan tipleri de tanımlanmaktadır. Yeni tanımlanan genlerle birlikte klinik ve genetik heterojenitesi giderek artmaktadır. Farklı genetik tipler arasında sıklıkla klinik olarak değişkenlik görülür. Ancak aynı genetik tiplerde farklı klinik veya farklı genetik tiplerde çok benzer klinikler ortaya çıkabilmektedir.

26
70’den fazla genetik HSP tip tanımlanmış olmasına rağmen sadece daha sık görülen birkaç tanesi klinik özellikler açısından iyi tanımlanmıştır. -SPG4, SPG3A, SPG11 Genetik tipler asrasında oldukça geniş klinik değişkenlik vardır. -Semptomların başlama yaşı, şiddeti, hastalığın seyri, güçsüzlüğün derecesi, diğer nörolojik bulguların görülmesi Değişkenliğin faklı genetik mutasyon dışında, altta yatan moleküler anormallikler ve modifiye edici faktörler (genetik/genetik olmayan) ile ilişkili olabileceği belirtilmektedir.
 ile ilişkili olabileceği belirtilmektedir..")
28
Spastisitenin ayırıcı tanısı; - semptomların başlama yaşı, - hastalığın seyri (statik/aylar içinde kötüşeme/yıllar içinde kötüleşme), -birinci derece akrabalarda görülme, -ek nörolojik sorunlar, HSP erken çocukluktan sonra başlar ve ilerleme sinsidir. Semptomların başlama zamanı net belirtiliyorsa HSP 6-12 ay içerisinde belirgin kötüleşme varsa olasılığı düşük

29
Tanıda zorluklar; - ailede benzer hastalık öyküsü olmaması ve genetik olmayan nedenler dışlanamaması - genetik test yaptıramama veya tamamlanmamsı, - eşlik eden durumların olması (diyabetik nöropati, servikal miyelopati, prematürüte) - anlamlı nörolojik veya nörogörüntüleme anormalliklerinin olması ( SSS ak madde patolojilerinin kanıtı) - semptom süresinin çok kısa olması ( < 5 yıl)
 - anlamlı nörolojik veya nörogörüntüleme anormalliklerinin olması ( SSS ak madde patolojilerinin kanıtı) - semptom süresinin çok kısa olması ( < 5 yıl)")
30
Tanı; Genetik testler, HSP klinik tanısını doğrulamak için en kullanışlı testlerdir. Test sonuçları, klinik temel ışığında yorumlanmaktadır. belirti ve bulguları HSP ile uyan, eşlik edebilecek veya alternatif tanıların dışladığı hastalarda, tanımlanan potansiyel patojenik HSP gen varyantları tanıyı doğrulayabilir.

31
Geniş panel yeni jenerasyon dizileme, birinci derece akrabalarda etkilenmiş aile öyküsü olan bireylerin büyük kısmında kesin tanıyı doğrulamaktadır. Ancak, HSP gen paneli yeni jenerasyon dizilemesi henüz tamamlanmamış; - tüm HSP genlerini içermiyor, - tüm kopya varyantlarını efektif olarak tanımlayamıyor

32
Praikte önerilen; Geniş panel HSP genleri yeni jenerasyon dizileme Dominant kalıtım paterni gösteren hastalarda SPG4 ekzon delesyon analizi (SPG3A ve SPG11) Diğer seçili HSP genleri için ekzon delesyon analizi (araştırma laboratuarları..)
 Diğer seçili HSP genleri için ekzon delesyon analizi (araştırma laboratuarları..)")
33
Genetik danışmanlık; - genetik tanısı kesinleşen ailelere, - prenatal tanı ve preimplantasyon genetik tanı hakkında bilgilendirme - tanımlanan genin, kalıtım paterni, penetrans derecesi

34
Yönetim Spesifik tedavi yok (koruma/durdurma/ geri döndürme) Hastaların büyük kısmında (hepsi değil) düzenli fiziksel egzersiz ile direnç artışı, yürüme becerisinde iyileşme ve ağrılarında azalma olmaktadır. Fizik tedavi uzmanı ve fizyoterapist konsültasyonu yapılmalı ve özel, her gün ve sürekli olacak şekilde bir egzersiz programı yapılmalıdır.

35
Güçlendirme (özellikle iliopsoas, hamstring ve tibialis anterior), gerginliği azaltma, denge ve hıza yönelik egzersizler yapılmalı. İlaçlar (lioresal, dantrolene); - po; güçsüzlüğü olmayan hastalarda - intratekal lioresal; deneme dozundan fayda - botilinum toksin; özellikle hamstring ve adduktor kaslar Üriner urgency tedavisi; oxybutinin Cerrahi
; - po; güçsüzlüğü olmayan hastalarda - intratekal lioresal; deneme dozundan fayda - botilinum toksin; özellikle hamstring ve adduktor kaslar Üriner urgency tedavisi; oxybutinin Cerrahi.")
36
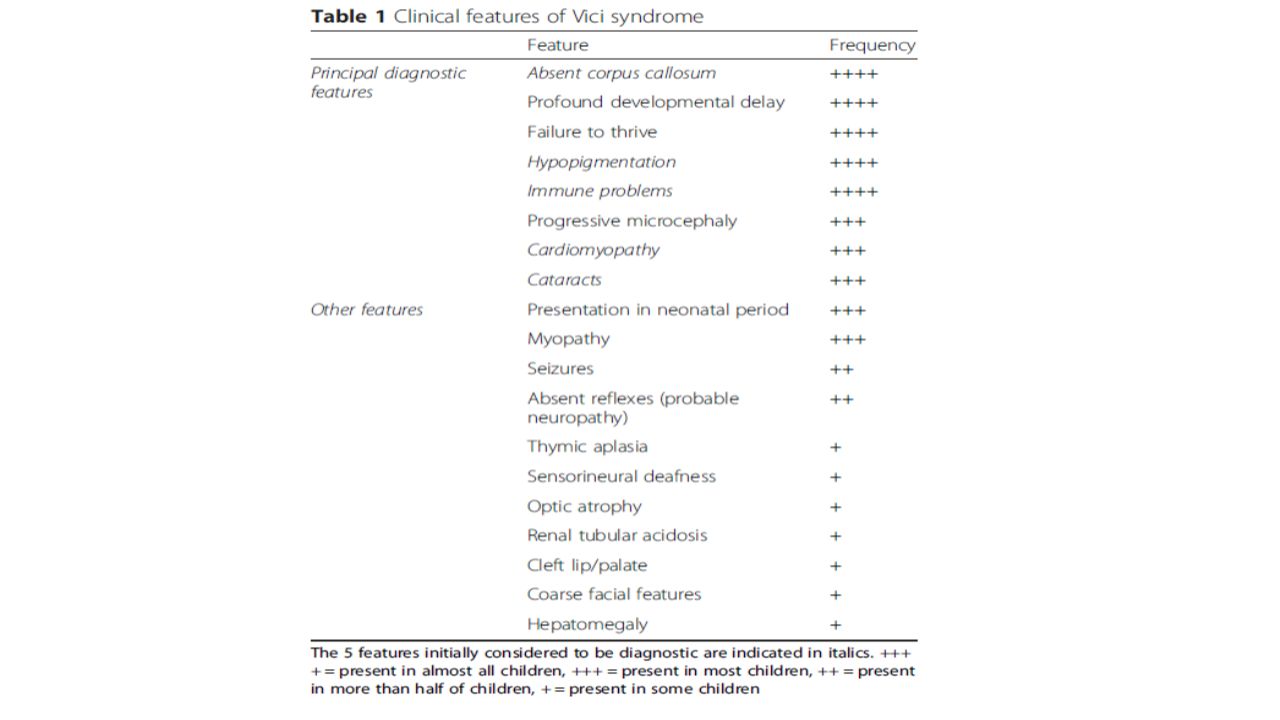
Vici Sendromu (Dianisi-Vici-Sabetta-Gambarara sendromu) İlk kez 1988 yılında Dianosi-Vici tarafından tanımlanmış; -İmmünyetmezlik ile birlikte yarık damak/dudak, katarakt, hipopigmentasyon ve korpus kallosum agenezisi İnsidans bilinmiyor, 50 civarında genetik olarak doğrulanmış vaka bildirilmiş. Çok nadir? Tanı konulamıyor?

37
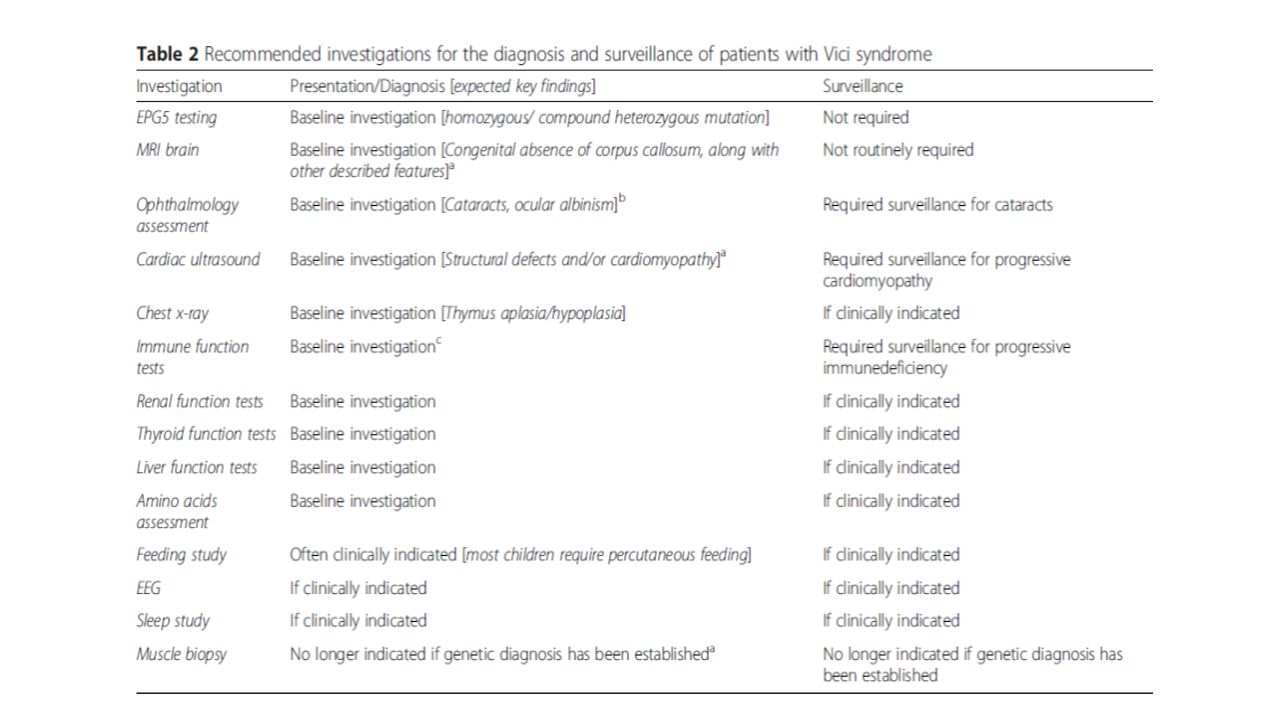
Tanım (OMIM/ORPHA); -18q12.3 kromozomunda yer alan EPG5 geninin OR mutasyonu (ektopik P granül protein 5 proteini kodlar) -Ağır konjenital multisistem tutulum -Temel özellikler; korpus kallozum agenezisi, katarakt, okulokutanöz hipopigmentasyon, kardiyomiyopati, kombine immün yetmezlik ve çok değişken multisistem tutulum Ağır gelişimsel gerilik, edinsel mikrosefali ve büyüme geriliği eşlik etmesi spesifik değil, ancak tanıyı yüksek oranda destekler.
; -18q12.3 kromozomunda yer alan EPG5 geninin OR mutasyonu (ektopik P granül protein 5 proteini kodlar) -Ağır konjenital multisistem tutulum -Temel özellikler; korpus kallozum agenezisi, katarakt, okulokutanöz hipopigmentasyon, kardiyomiyopati, kombine immün yetmezlik ve çok değişken multisistem tutulum Ağır gelişimsel gerilik, edinsel mikrosefali ve büyüme geriliği eşlik etmesi spesifik değil, ancak tanıyı yüksek oranda destekler.")
39
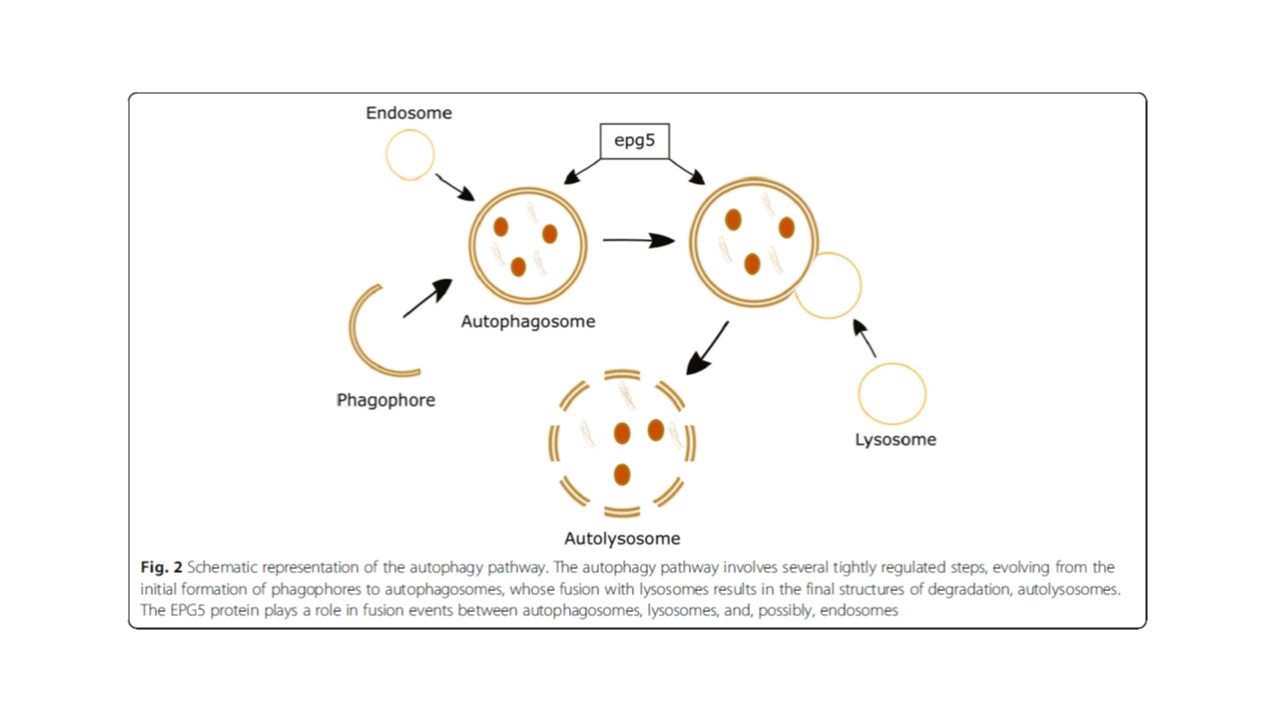
EPG5 proteini çok hücreli organizmalarda otofajiyi düzenlemeye yönelik çok önemli bir role sahiptir. Otofaji, bozulmuş/işlevi olmayan proteinlerin ve organellerin uzaklaştırılmasında önemli rolü olan, evrim boyunca korunmuş temel bir hücresel indirgeyici yoldur. Enfeksiyon yanıtı, metabolik yolaklar… Otofaji, nöronlar ve kasta fizyolojik olarak vardır ve hastalığın belirgin santral sinir sistemi ve nöromüsküler tutulumunun bununla ilgili olduğu düşünülüyor. Santral sinir sisteminde patolojik ve klinik olarak nörodejeneratif, ileyleyici bir tabloya nden olmaktadır.

42
Teşekkürler..

Benzer bir sunumlar