Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARINA YAKLAŞIM
Dr. Şule Şengül Nefroloji Bilim Dalı

2
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI
Tüm hastalıklarda bilinen veya henüz bilemediğimiz genetik bir bileşen vardır Böbreklerde değişen derecelerde yapısal ve/veya fonksiyonel bozukluklara yol açabilirler Diğer sistemlerle ilgili bulgular eşlik edebilir

3
Kronik Böbrek Hastalığı (KBH): RİSK FAKTÖRLERİ
Klinik DM HT KBY aile öyküsü Otoimmün hastalıklar Sistemik infeksiyonlar Üriner infeksiyon/Taş hastalığı Alt üriner trakt obstrüksiyonu Malignite Geçirilmiş ABY İlaçlar Azalmış renal kitle Düşük doğum ağırlığı Sosyodemografik İleri yaş (>60) Irk Kimyasal/çevresel maruziyet Düşük gelir/eğitim
 Irk. Kimyasal/çevresel maruziyet. Düşük gelir/eğitim.")
4
Filtrasyon fonksiyonunu etkileyen global hastalıklar
KBH:Etiyoloji DM HT Ateroskleroz Glomerüler hastalıklar Toksik maddeler İlaçlar Kalıtsal hastalıklar Tübüler hastalıklar Vasküler hastalıklar Transplant nefropatisi Filtrasyon fonksiyonunu etkileyen global hastalıklar

5
Kidney disorder or syndrome Genes Proteins/Products
Alport syndrome (X linked) COL4A5 Type IV collagen α5 chain Alport Syndrome (autosomal recessive) COL4A3 or COL4A4 Type IV collagen α3 chain Type IV collagen α4 chain Alport syndrome with leiomyomatosis (X linked) COL4A5 and COL4A6 Type IV collagen α5 and α6 chain Benign familial hematuria (autosomal dominant) COL4A4 Type IV collagen α4 chain Autosomal dominant polycystic kidney disease 1 (PKD1) PKD1 Polycystin 1 Autosomal dominant polycystic kidney disease 2 (PKD2) PKD2 Polycystin 2 Autosomal recessive PKD PKD3 Polycystin ? VonLippel-Lindau (VHL) disease TSC/VHL VHL protein Nephrogenic diabetes insipidus (X- linked) ADHRV2 Vasopresin receptor V2 Nephrogenic diabetes insipidus (autosomal recessive) AQP2 Aquaporin 2 Familial hypocalcuric hypercalcemia CASR Ca 2+ sensing receptor X- linked recessive nephrolithiasis CLCN5 Cl- channel X- linked recessive hypophosphatemic rickets Fabry disease (X- linked) GLA α-galactosidaseA (α-galA) Juveline nephronophtysis NPHP1 Steroid resistant nephrotic syndrome NPHS2 podocin
 COL4A5. Type IV collagen α5 chain. Alport Syndrome (autosomal recessive) COL4A3 or COL4A4. Type IV collagen α3 chain Type IV collagen α4 chain. Alport syndrome with leiomyomatosis (X linked) COL4A5 and COL4A6. Type IV collagen α5 and α6 chain. Benign familial hematuria (autosomal dominant) COL4A4. Type IV collagen α4 chain. Autosomal dominant polycystic kidney disease 1 (PKD1) PKD1. Polycystin 1. Autosomal dominant polycystic kidney disease 2 (PKD2) PKD2. Polycystin 2. Autosomal recessive PKD. PKD3. Polycystin VonLippel-Lindau (VHL) disease. TSC/VHL. VHL protein. Nephrogenic diabetes insipidus (X- linked) ADHRV2. Vasopresin receptor V2. Nephrogenic diabetes insipidus (autosomal recessive) AQP2. Aquaporin 2. Familial hypocalcuric hypercalcemia. CASR. Ca 2+ sensing receptor. X- linked recessive nephrolithiasis. CLCN5. Cl- channel. X- linked recessive hypophosphatemic rickets. Fabry disease (X- linked) GLA. α-galactosidaseA (α-galA) Juveline nephronophtysis. NPHP1. Steroid resistant nephrotic syndrome. NPHS2. podocin.")
6
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

7
Kistik Böbrek Hastalıkları
Genetik geçişliler Otozomal dominant (OD) OD polikistik böbrek hastalığı Von Hippel-Lindau sendromu Tuberoz sklerozis Erişkin başlangıçlı medüller kistik hastalık Otozomal resesif (OR) OR polikistik böbrek hastalığı Juvenil nefronofitizis Meckel-Gruber sendromu X-bağımlı Orofasiyaldigital sendrom tip I Genetik olmayanlar Gelişimsel Medüller sünger böbrek Renal kistik displazi Multikistik displazi Alt üriner sistem obs. İle kistik displazi Diffüz kistik displazi Sendromal Non-sendromal Akkiz Basit kistler Hipokalemik kistik hastalık Akkiz kistik böbrek hastalığı
 OD polikistik böbrek hastalığı. Von Hippel-Lindau sendromu. Tuberoz sklerozis. Erişkin başlangıçlı medüller kistik hastalık. Otozomal resesif (OR) OR polikistik böbrek hastalığı. Juvenil nefronofitizis. Meckel-Gruber sendromu. X-bağımlı. Orofasiyaldigital sendrom tip I. Genetik olmayanlar. Gelişimsel. Medüller sünger böbrek. Renal kistik displazi. Multikistik displazi. Alt üriner sistem obs. İle kistik displazi. Diffüz kistik displazi. Sendromal. Non-sendromal. Akkiz. Basit kistler. Hipokalemik kistik hastalık. Akkiz kistik böbrek hastalığı.")
9
ADPKD özellikleri Multisistemik bir hastalık
Böbrek, KC, pankreas, araknoid membranlarda kistler Kistik olmayan ekstrarenal bulgular MVP İntrakranial anevrizmalar Herniler.... Sıklık 1/ En yaygın genetik böbrek hastalığıdır. Avrupa ve Amerika’da son dönem böbrek yetersizliklerinin yaklaşık % 5’inden sorumludur. Otozomal dominant Genetik heterojenite Semptomlar orta yaşlarda çıkar Büyük polikistik böbrekler

10
APKD Genetik Heterojenite

12
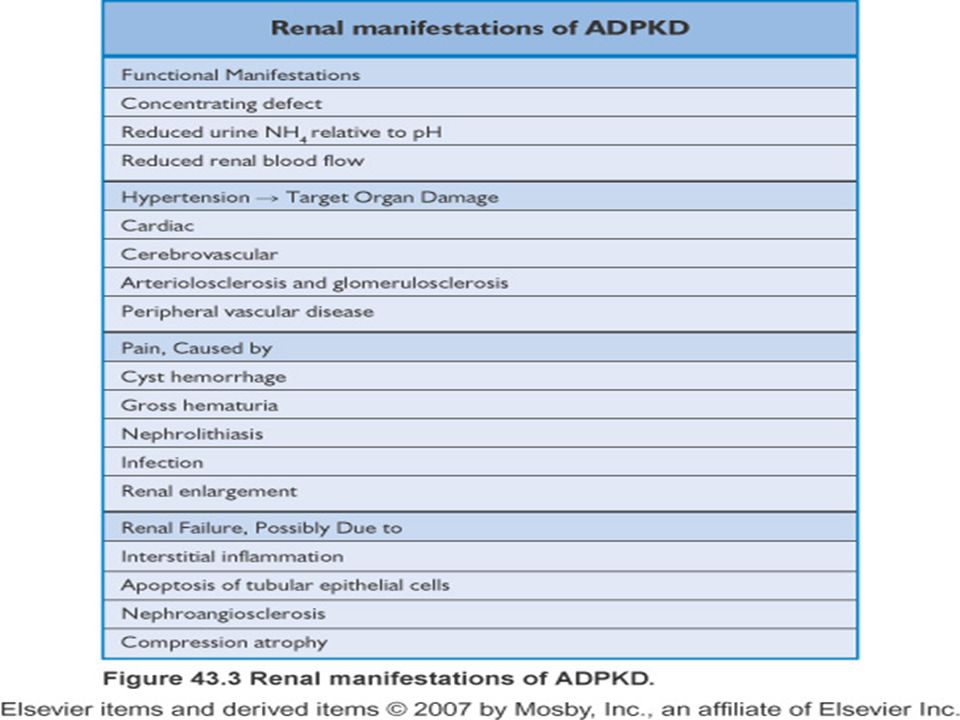
ADPKBH:Renal Bulgular
Böbreklerde büyüme sonuçta %100 Hematüri ve hemoraji %40 Nefrolitiazis %20 Hipertansiyon böbrek yetmezliği gelişmeden önce %75 gelişir ve yaşla birlikte artar Son dönem böbrek yetmezliği yaşları arasında %50 gelişir

14
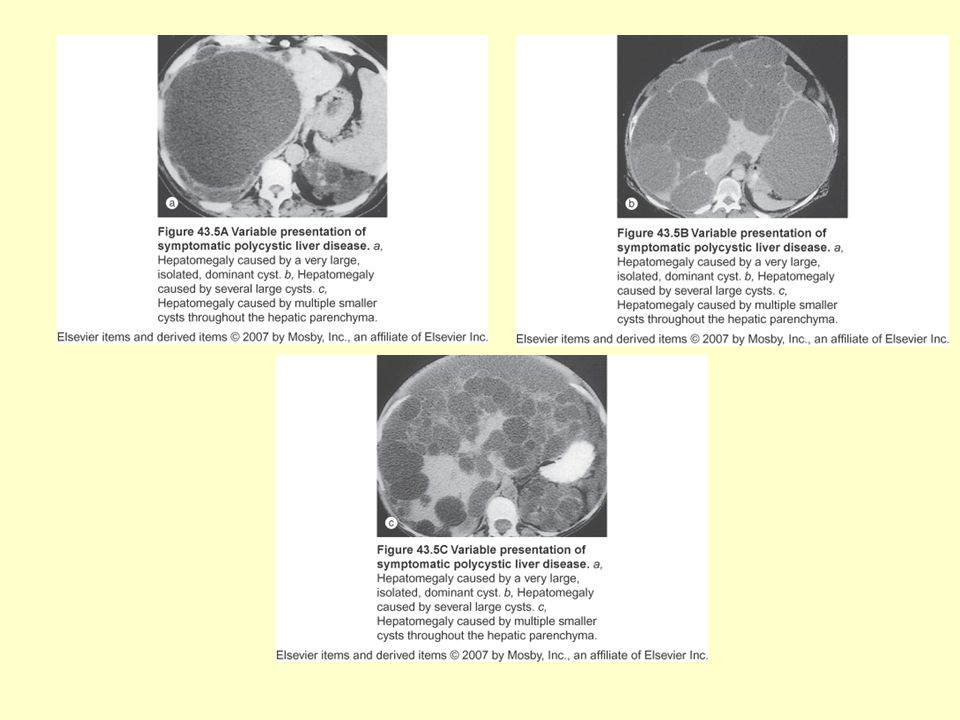
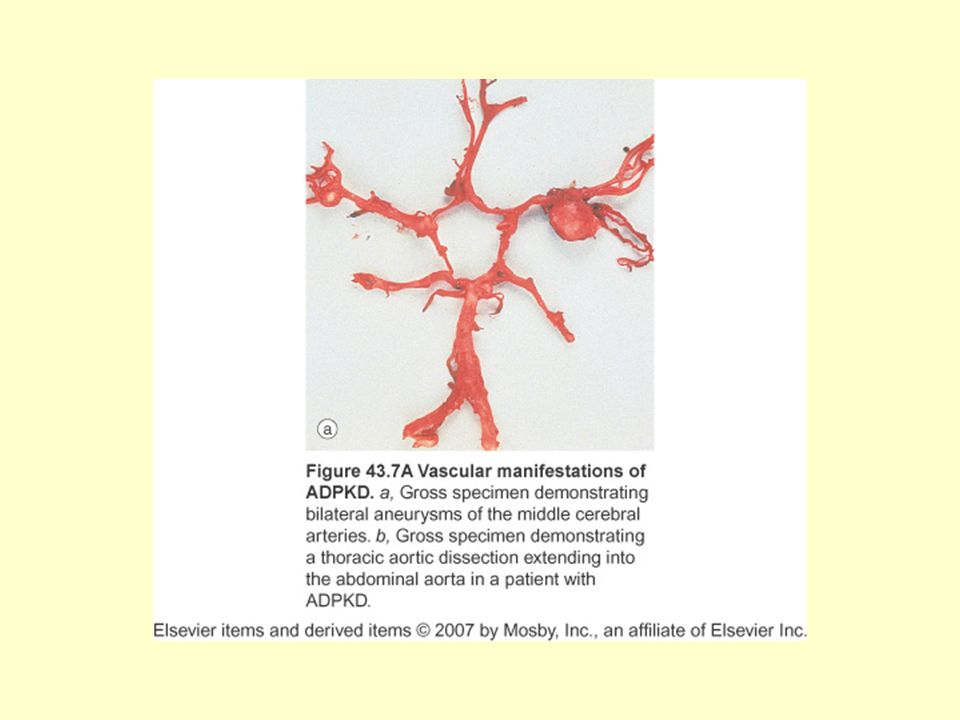
ADPKBH:Ekstrarenal Bulgular
Polikistik KC hastalığı: En sık ekstrarenal bulgudur İntrakraniyal anevrizma: %8 Koroner arter anevrizmaları Torasik aorta diseksiyonu Cervikosefalik arteriyel diseksiyon Valvular kalp hastalığı: MVP %25

17
ADPKBH: Tanı Ultrasonografi Yaş Aile hikayesi pozitif genotip? <40
En az 3 uni ya da bilateral kist 40-59 En az 2 kist her bir böbrekte ≥60 En az 4 kist her bir böbrekte Yaş Genotip 1+ <30 En az 2 uni ya da bilateral kist 30-59 2 kist her bir böbrekte ≥ 60 4 kist her bir böbrekte

18
Renal tümörler ile ilgili genetik kistik hastalıklar
Von Hippel-Lindau (VHL) sendromu Otozomal dominant geçişli VHL gen tümör supresör etkilidir 3q25 yerleşimindeki gende mutasyonlar saptanabilir Tuberous Sclerosis Complex (TSC) TSC1 9q protein: Hamartin TSC 2 16p13 protein: Tuberin Ailevi Renal Hücreli Karsinom Ailevi renal hücreli karsinomlarda kromozom 3 translokasyonu saptanmıştır. Translokasyon sonucu DIRC2 (disrupted in renal cancer) geni oluşmaktadır
 sendromu. Otozomal dominant geçişli. VHL gen tümör supresör etkilidir. 3q25 yerleşimindeki gende mutasyonlar saptanabilir. Tuberous Sclerosis Complex (TSC) TSC1 9q32.34 protein: Hamartin. TSC 2 16p13 protein: Tuberin. Ailevi Renal Hücreli Karsinom. Ailevi renal hücreli karsinomlarda kromozom 3 translokasyonu saptanmıştır. Translokasyon sonucu DIRC2 (disrupted in renal cancer) geni oluşmaktadır.")
19
Renal tümörler ile ilgili genetik kistik hastalıklar von Hippel-Lindau (VHL) sendromu
Göz (hemangioblastoma) Serebellum (hemangioblastoma) Spinal kord Adrenal gland Pankreas Epididimis Renal ve pankreatik kistler 1:36.000 Tip I TipIIA TipIIB TipIIC
 Serebellum (hemangioblastoma) Spinal kord. Adrenal gland. Pankreas. Epididimis. Renal ve pankreatik kistler. 1: Tip I. TipIIA. TipIIB. TipIIC.")
20
Renal tümörler ile ilgili genetik kistik hastalıklar Tuberous Sclerosis Complex (TSC)
Hamartomlar Böbrekler (angiomyolipomlar ve kistler) Beyin Kalp Akciğerler Cilt 1:6000
 Beyin. Kalp. Akciğerler. Cilt. 1:6000.")
21
Medüller Kistik Böbrek Hastalığı: MCKD
Otozomal dominant SDBY yaşlarında gelişir Proteinüri -/minimal Medüller kistler USG de görülebilir MCKD Tip 1 MCKD Tip 2: Gut ve hiperürisemi (familiyal juvenil hiperürisemik nefropati)
")
22
MCKD Tip 1 Yavaş ilerleyen böbrek yetmezliğine yol açar
Normal idrar incelemesi, hafif proteinüri veya mikroskopik hematüri görülebilir Hiperürisemi ve gut nadir eşlik eder, böbrek yetmezliğinin ileri evrelerinde görülebilir Aile öyküsünün alınabilmesi çok önemlidir Hastaların %40’ında USG de kistler görülebilir Renal biyopside; tübüler atrofi, intersitisyel fibrozis, tübüler basal membranda karakteristik lamellasyon Spesifik bir tedavisi yoktur

23
MCKD Tip 2 (Familiyal Juvenil Hiperürisemik Nefropati)
Mutasyon üromodulin genindedir (UMOD geni) Hastalığın erken dönemlerinden itibaren (<20 yaş)hiperürisemi ve gut gözlenir Progressif böbrek yetmezliğine yol açar Mutant uromodulin tübüllerde birikir Böbrek biyopsisinde tübülointersitisyel hastalık saptanır, ürat kristalleri gözlenmez Tanıda ailede böbrek yetmezliği ve gut hikayesi, normale yakın idrar bulguları saptanması önemlidir USG bulgusu genellikle saptanmaz Mutasyon analizi kesin tanı için yapılabilir
 Hastalığın erken dönemlerinden itibaren (<20 yaş)hiperürisemi ve gut gözlenir. Progressif böbrek yetmezliğine yol açar. Mutant uromodulin tübüllerde birikir. Böbrek biyopsisinde tübülointersitisyel hastalık saptanır, ürat kristalleri gözlenmez. Tanıda ailede böbrek yetmezliği ve gut hikayesi, normale yakın idrar bulguları saptanması önemlidir. USG bulgusu genellikle saptanmaz. Mutasyon analizi kesin tanı için yapılabilir.")
24
Kistik Böbrek Hastalıkları
Genetik geçişliler Otozomal dominant (OD) OD polikistik böbrek hastalığı Erişkin başlangıçlı medüller kistik hastalık Von Hippel-Lindau sendromu Tuberoz sklerozis Otozomal resesif (OR) OR polikistik böbrek hastalığı Juvenil nefronofitizis Meckel-Gruber sendromu X-bağımlı Orofasiyaldigital sendrom tipI Genetik olmayanlar Gelişimsel Medüller sünger böbrek Renal kistik displazi Multikistik displazi Alt üriner sistem obs. İle kistik displazi Diffüz kistik displazi Sendromal Non-sendromal Akkiz Basit kistler Hipokalemik kistik hastalık Akkiz kistik böbrek hastalığı
 OD polikistik böbrek hastalığı. Erişkin başlangıçlı medüller kistik hastalık. Von Hippel-Lindau sendromu. Tuberoz sklerozis. Otozomal resesif (OR) OR polikistik böbrek hastalığı. Juvenil nefronofitizis. Meckel-Gruber sendromu. X-bağımlı. Orofasiyaldigital sendrom tipI. Genetik olmayanlar. Gelişimsel. Medüller sünger böbrek. Renal kistik displazi. Multikistik displazi. Alt üriner sistem obs. İle kistik displazi. Diffüz kistik displazi. Sendromal. Non-sendromal. Akkiz. Basit kistler. Hipokalemik kistik hastalık. Akkiz kistik böbrek hastalığı.")
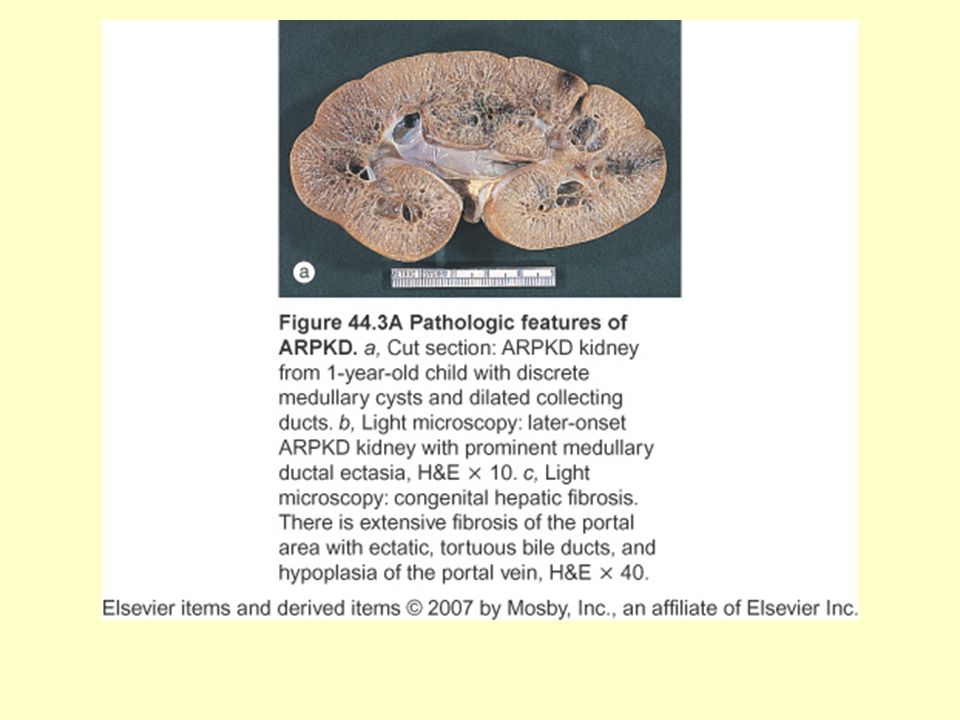
25
ARPKD Renal toplayıcı kanallarda dilatasyon ve biliyer duktal ektaziyle karakterizedir 1:20000 (canlı doğumda) 1994 (Gen lokus 6p12.1-p21) 2002 (Ward ve ark) PKHD1 klonlandı Hastaların çoğu inutero veya doğumda tanınır Ekojenik böbrekler, oligohidroamniyoz saptanır Potter fenotipi: pulmoner hipoplazi+ekstremite spinal anomaliler+tipik yüz görünümü Hipertansiyon %70-80 ilk aylardan itibaren görülür Hematüri, proteinüri, steril piyüri
 2002 (Ward ve ark) PKHD1 klonlandı. Hastaların çoğu inutero veya doğumda tanınır. Ekojenik böbrekler, oligohidroamniyoz saptanır. Potter fenotipi: pulmoner hipoplazi+ekstremite spinal anomaliler+tipik yüz görünümü. Hipertansiyon %70-80 ilk aylardan itibaren görülür. Hematüri, proteinüri, steril piyüri.")
26
ARPKD Prenatal tanıda fetal USG, genetik test yapılabilir
Postnatal tanıda görüntüleme yöntemleri kullanılabilir Perinatal mortalite %30-50 Kombine Kc ve böbrek nakli gerekebilir

28
Juvenil Nefronofitizis
Klinik presantasyon Gen Exons Protein Juvenil form NPHP1 (2q12-13) NPHP4 (1p36) 20 30 nephrocystin nephrocystin-4/ nephroretinin Adolesan form Senior-Løken sendromu NPHP3 (3q21-q22) NPHP5 (3q) IQCB1 27 13 nephrocystin-3 nephrocystin-5 Infantil form NPHP2 (9q22-31) INVS 16 inversin
 NPHP4 (1p36) nephrocystin. nephrocystin-4/ nephroretinin. Adolesan form. Senior-Løken. sendromu. NPHP3 (3q21-q22) NPHP5 (3q) IQCB nephrocystin-3. nephrocystin-5. Infantil form. NPHP2 (9q22-31) INVS. 16. inversin.")
29
Juvenil Nefronofitizis
Çocuk ve adolesanlarda SDBY vakalarının %6-15’ini oluşturur Hastalığın başlangıç yaşına göre: İnfantil Juvenil Adolesan İdrar konsantrasyon defekti Poliüri/polidipsi Böbrek yetmezliği Gelişme geriliği Retinitis pigmentosa, retinal distrofi, nistagmus

30
Medüller Sünger Böbrek
Medüller ve papiller bölgedeki toplayıcı kanallarda dilatasyonla karakterizedir (sünger görünümü) Konjenital hemihipertrofi, Beckwith-Wiedeman sendromu, Ehler-Danlos sendromu ve Marfan sendromu gibi durumlara eşlik edebilen gelişimsel bir bozukluktur Sıklık 1:5000 Genellikle asemptomatiktir Taş hastalığı, hematüri ve infeksiyonla ortaya çıkabilir Böbrek yetmezliğine progresyon göstermesi son derece nadirdir
 Konjenital hemihipertrofi, Beckwith-Wiedeman sendromu, Ehler-Danlos sendromu ve Marfan sendromu gibi durumlara eşlik edebilen gelişimsel bir bozukluktur. Sıklık 1:5000. Genellikle asemptomatiktir. Taş hastalığı, hematüri ve infeksiyonla ortaya çıkabilir. Böbrek yetmezliğine progresyon göstermesi son derece nadirdir.")
31
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

32
Genetik Glomerüler Sendromlar
Alport sendromu İnce basal membran hastalığı Tırnak-patella sendromu Ailesel fokal segmentalglomerüloskleroz Ailesel Ig A nefropatisi

33
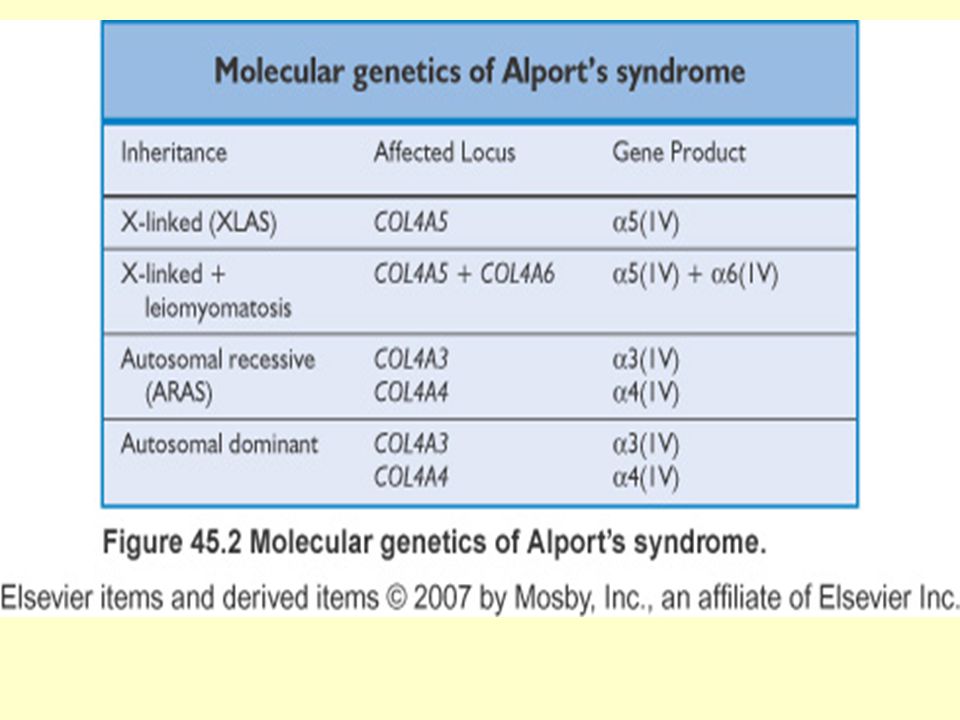
Alport Sendromu: (AS) Basal membrandaki tipIV kollagenin spesifik proteinlerini (alfa 3-5 zincirleri) etkileyen mutasyonlar sözkonusudur Başlıca bulgular: Hematüri Proteinüri ile beraber progressif nefrit Böbrek yetmezliği Sensörinöral sağırlık Okuler anormallikler (anterior lentikonus-patognomonik bulgu-, makulopati) Sıklık = 1/5000 X-bağımlı AS vakaların %80 ini oluşturur Kardinal bulgu HEMATÜRİDİR.
 Sıklık = 1/5000. X-bağımlı AS vakaların %80 ini oluşturur. Kardinal bulgu HEMATÜRİDİR.")
35
İnce Basal Membran Hastalığı
Benign familiyal hematüri Genel populasyonda %5-9 COL4A3 ve COL4A4 genlerinde heterozigot defektler söz konusudur Otozomal dominant Aile öyküsü % Mikroskopik hematüri, makroskopik hematüri, yan ağrısı görülebilir Proteinüri ve hipertansiyon nadirdir Uzun dönem prognoz genellikle iyidir

36
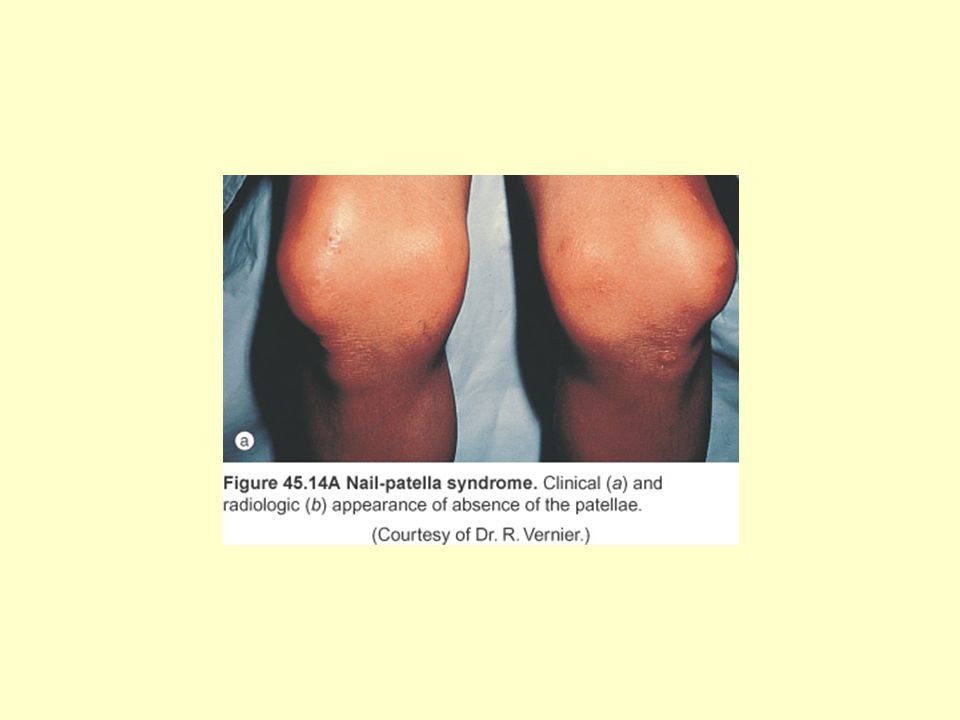
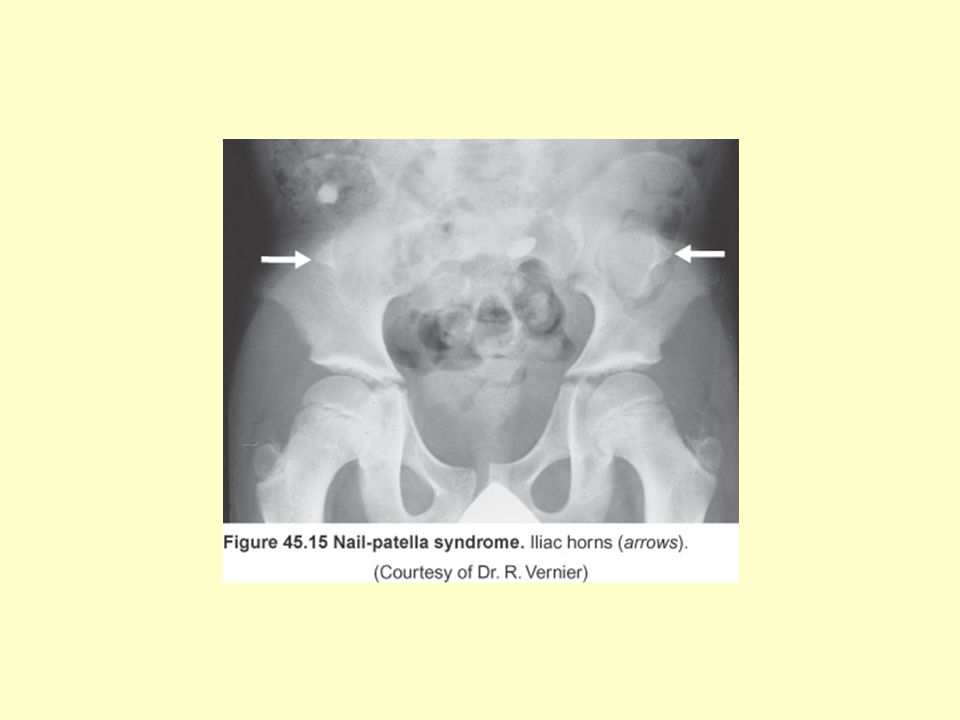
Tırnak-Patella Sendromu: (Osteoonikodisplazi)
Otozomal dominant (kromozom 9) Patella yokluğu veya hipoplazisi Distrofik tırnaklar Bileklerde ve iliak kanatlarda displazi 22: (insidans) Renal bulgular (proteinüri, hematüri, nefrotik sendrom, hipertansiyon) hastaların %50’sinde görülür Ortalama 33 yaşında hastaların %30’unda son dönem böbrek yetmezliği gelişir
 Patella yokluğu veya hipoplazisi. Distrofik tırnaklar. Bileklerde ve iliak kanatlarda displazi. 22: (insidans) Renal bulgular (proteinüri, hematüri, nefrotik sendrom, hipertansiyon) hastaların %50’sinde görülür. Ortalama 33 yaşında hastaların %30’unda son dönem böbrek yetmezliği gelişir.")
40
Ailesel Nefrotik Sendromlar/FSGS
Otozomal resesif kalıtım Fin tipi konjenital NS (NPHS1- 19q13) Steroid-resistan NS (NPHS2-1q25-31) Pierson sd (LAMB2 - 3p14-p22) – (mikrokoria-konj NS) Otozomal dominant kalıtım FSGS-1 locus: krom 19q13 (ACTN4) FSGS-2 locus: krom 11q21-q22 (TRPC6) FSGS-3 locus : krom 6 (CD2-associated protein) WT1 Epstein sd : MYH9 (non muscle myosin heavy chain IIA) Mitokondriyal sitopati ile birlikte (MELAS sendromu) Genellikle sağırlık ve/veya diyabet ve/veya diğer eksrarenal hastalıklar ile, ancak proteinüri ilk semptom olabilir. En sık mutasyon: A324G
 Steroid-resistan NS (NPHS2-1q25-31) Pierson sd (LAMB2 - 3p14-p22) – (mikrokoria-konj NS) Otozomal dominant kalıtım. FSGS-1 locus: krom 19q13 (ACTN4) FSGS-2 locus: krom 11q21-q22 (TRPC6) FSGS-3 locus : krom 6 (CD2-associated protein) WT1. Epstein sd : MYH9 (non muscle myosin heavy chain IIA) Mitokondriyal sitopati ile birlikte (MELAS sendromu) Genellikle sağırlık ve/veya diyabet ve/veya diğer eksrarenal hastalıklar ile, ancak proteinüri ilk semptom olabilir. En sık mutasyon: A324G.")
41
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

42
Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar
Fabry hastalığı Sistinoz Hiperoksalüri

43
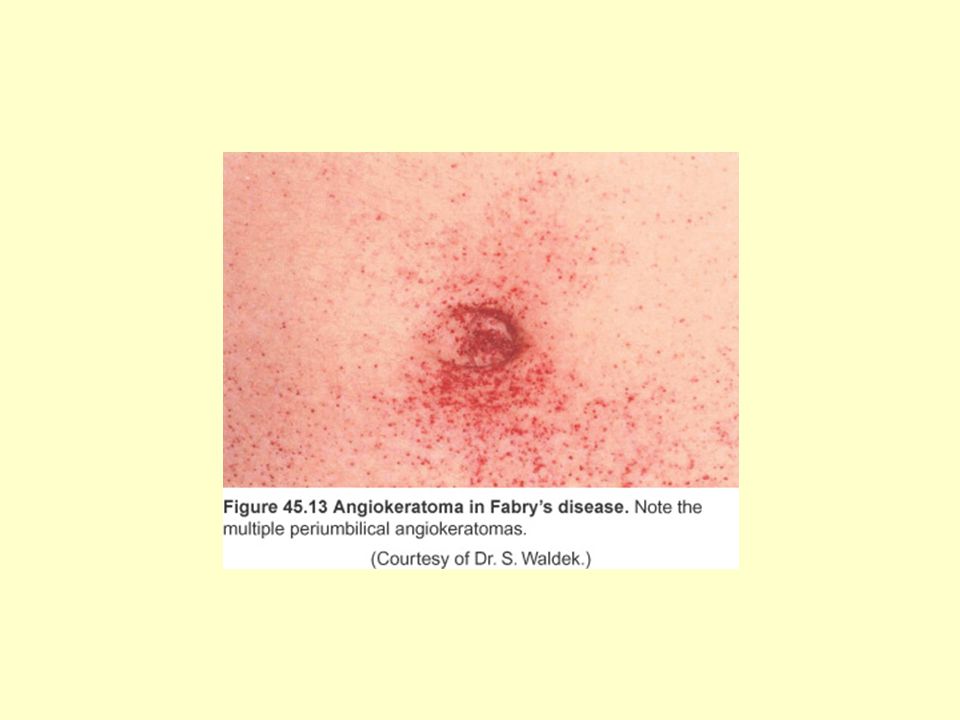
Tanı renal biyopsi ile yapılır rekombinant -galactosidase A
Fabry Hastalığı -galactosidase A eksikliğine bağlı X-bağlı geçişli Akroparestezi, angiokeratoma, hipohidrozis ya da korneal/lenticular lezyonlar Renal, kardiovasküler ve serebral hastalık Hasta gen Xq22.1 üzerinde yer alır İzole renal hastalık: Proteinüri ve yüksek kan basıncı Tanı renal biyopsi ile yapılır 200 üzerinde mutasyon Prenatal tanı + rekombinant -galactosidase A

45
Sistinoz Sistinoz: otozomal resesif geçişli, CTNS geninde (17p13) de mutasyon sonucu lizozomlarda sistin birikimi İnsidans ~ 1 : 200,000 yenidoğanda Tüm dünyada ~ 2000 sistinozlu hasta İnfantil nefropatik, juvenil, adolesan tipler ilk 9 eksonda 65 kb bir delesyon Hızlı bir PCR yöntem gelişmiştir

46
Primer Hiperoksalüri PH1 PH2 AGXT gen defekti
Glyoxylate aminotransferase enzim eksikliği Enzim analizi ile tanı konulan olguların ancak % 47’sinde mutasyon gösterilmiş. Prenatal tanı ve taşıyıcılık açısından değerli Kromozom bölge: 2q36-37 PH2 GRHPR gen defekti Glyoxylate reductase/hydroxypyruvate reductase enzim eksikliği Gen sembolü: GRHPR Kromozom bölge: 9cen

47
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

48
Genetik Tübüler Bozukluklar
Ailesel glukoz-galaktoz malabsopsiyonu Herediter renal glikozüri Aminoasidüriler Sistinüri Hartnup hastalığı İminoglisinüri Lisinürik protein intoleransı Herediter ürik asit tutulum bozuklukları Renal hipoürisemi Familiyal juvenil hiperürisemik nefropati Fanconi sendromunun kalıtsal nedenleri Sistinozis Galaktozemi Fruktoz intoleransı Tirozinemi Wilson hastalığı Lowe sendromu (okuloserebrorenal sendrom) Dent hastalığı
 Dent hastalığı.")
49
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

50
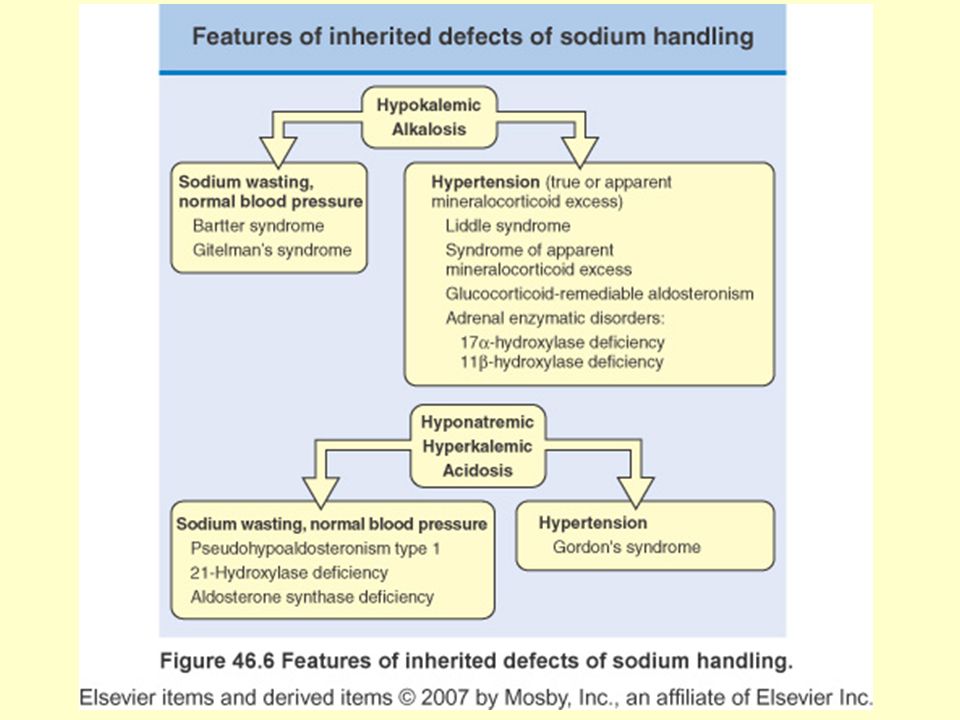
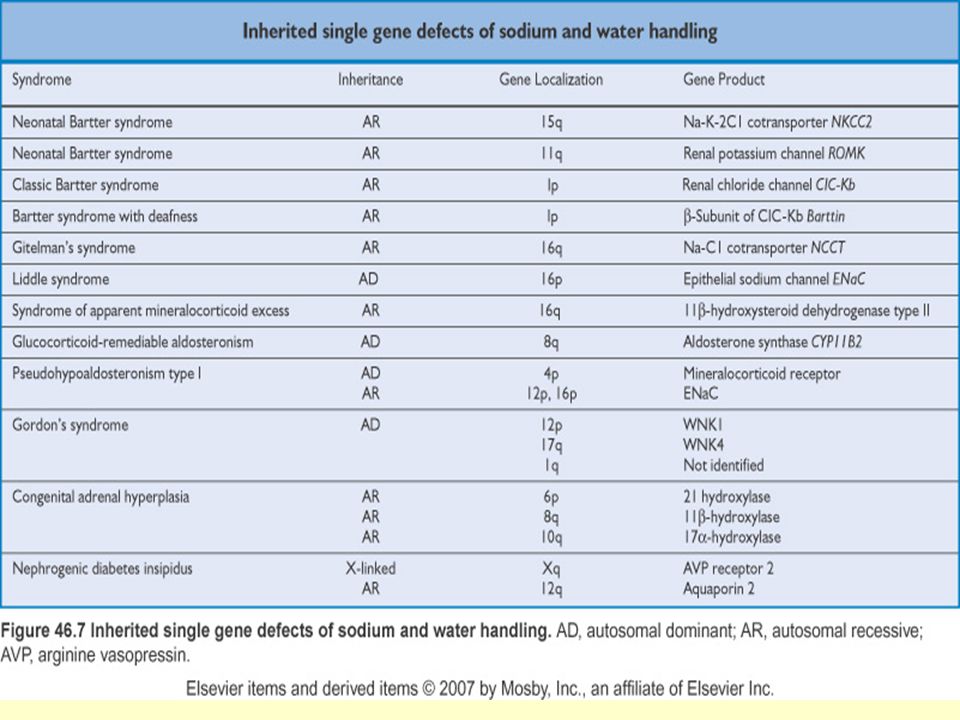
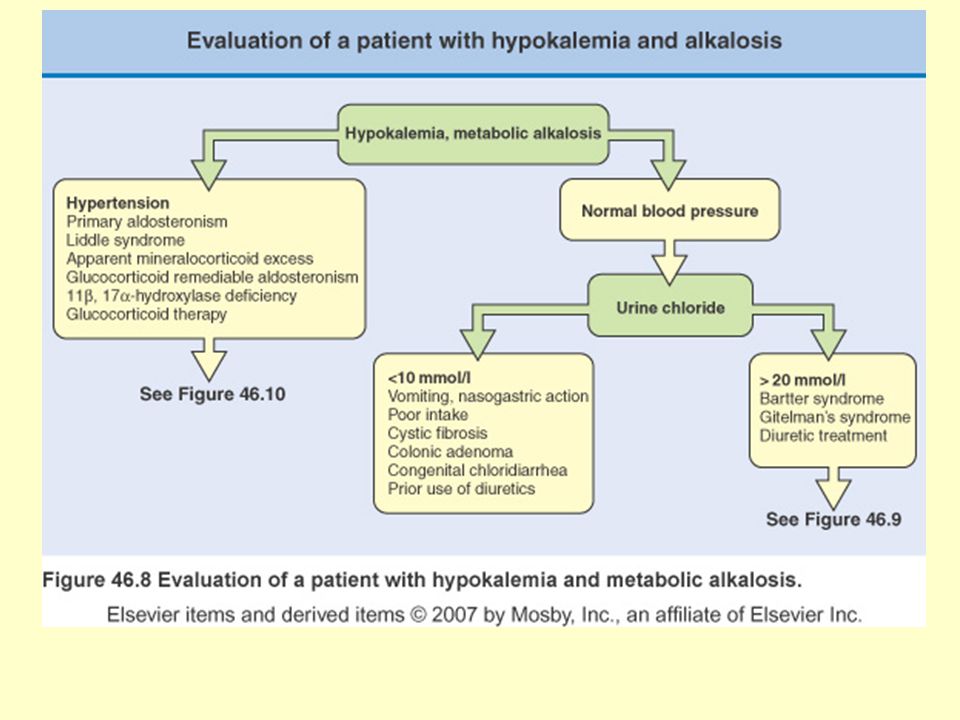
Ailesel Su ve Sodyum Metabolizması Bozuklukları
Bartter sendromu Gitelman sendromu Liddle sendromu Mineralokortikoid fazlalığı Glukokortikoidle düzeltilebili aldosteronizm Adrenal enzim eksiklikleri 17 alfa-hidroksilaz eksikliği 11 beta-hidroksilaz eksikliği Psödohipoaldosteronizm tip 1 21-hidroksilaz eksikliği Aldosteron sentaz eksikliği Gordon sendromu Konjenital nefrojenik diabetes insipitus

54
Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar
HEREDİTER VE KONGENİTAL BÖBREK HASTALIKLARI Kistik böbrek hastalıkları Genetik proteinürik / hematürik hastalıklar Böbrek ile ilgili lizozomal ve peroksizomal hastalıklar Genetik tubüler hastalıklar Ailesel su ve sodyum metabolizması bozuklukları Genetik geçişli trombotik mikroanjiopatiler

55
Genetik geçişli trombotik mikroanjiopatiler
Hemolitik Üremik Sendrom (HUS) C3 konvertaz aktivitesini kontrol eden faktör H fonksiyonunda azalma HUS ve MPGN II oluşumunda rol oynar. Ailevi ve sporadik HUS vakalarında faktör H mutasyonları gösterilmiştir. Trombotik Trombositopenik Purpura (TTP) Von Willebrand-cleaving protease ADAMTS13 eksikliği TTP oluşturur. Mutasyon 9q34 bölgesinde ADAMTS13 genindedir.
 C3 konvertaz aktivitesini kontrol eden faktör H fonksiyonunda azalma HUS ve MPGN II oluşumunda rol oynar. Ailevi ve sporadik HUS vakalarında faktör H mutasyonları gösterilmiştir. Trombotik Trombositopenik Purpura (TTP) Von Willebrand-cleaving protease ADAMTS13 eksikliği TTP oluşturur. Mutasyon 9q34 bölgesinde ADAMTS13 genindedir.")
Benzer bir sunumlar