Sunuyu indir
1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
Anabilim Dalı Hematoloji Bilim Dalı Olgu Sunumu 6 Mart 2012

2
ÇOCUK HEMATOLOJİ VAKA SUNUMU
MART 2012 ÇOCUK HEMATOLOJİ VAKA SUNUMU

3
23.01.2012 54 günlük, erkek hasta Şikayeti: kilo alamama
Hikaye : 40 gh’da NSVY ile 3600 g doğan hasta 27 günlükken aşırı kilo kaybetmesi (% 13,5) nedeniyle YDYBU’ne yatırılmış. İdrar yolu infeksiyonu tanısı almış. Böbrek USG: grade I pelviektazi saptanmış. Tedavi sonrası ayaktan izleme alınmış. YDYBU kontrol polikliniğine geldiğinde 2,8 g/gün aldığı tespit edilmiş.
 nedeniyle YDYBU’ne yatırılmış. İdrar yolu infeksiyonu tanısı almış. Böbrek USG: grade I pelviektazi saptanmış. Tedavi sonrası ayaktan izleme alınmış. YDYBU kontrol polikliniğine geldiğinde 2,8 g/gün aldığı tespit edilmiş.")
4
23. 01. 2012 Özgeçmiş: 4. gebelikten 40 gh’da NSVY ile doğmuş
Özgeçmiş: 4. gebelikten 40 gh’da NSVY ile doğmuş. Prenatal takibinde problem yok. Doğum sırasında müdahele yok. İkter, siyanoz gelişmemiş. İlk 1 ay anne sütü, sonrasında ek olarak formül ile beslenmeye başlanmış. Soygeçmiş: anne__31 yaş, sağ/sağlıklı, ev hanımı, ilkokul, 4 gebeliğinden biri 38. haftada intrauterin ex baba__36 yaş, sağ/sağlıklı, işçi, ilkokul 8 yaş, erkek ve 4,5 yaş kız sağ sağlıklı kardeş Anne ve baba uzaktan akraba!!

5
Fizik Muayene Genel durum iyi ,
3750 g (<3p), BÇ:36,5 cm (10p), Boy:52 cm (<3p) Cilt kuru, gövdede basmakla solan kırmızı renkli maküler döküntü mevcut Baş boyun: Saç ve saçlı deri doğal. Kafa yapısı simetrik. Ön fontanel 3X3 cm Gözler: Işık refleksi bilateral mevcut. Pupiller izokorik. Kardiyovasküler: KTA:120/dk, S1, S2 doğal. S3 yok. Üfürüm yok, AFN +/+
, BÇ:36,5 cm (10p), Boy:52 cm (<3p) Cilt kuru, gövdede basmakla solan kırmızı renkli maküler döküntü mevcut. Baş boyun: Saç ve saçlı deri doğal. Kafa yapısı simetrik. Ön fontanel 3X3 cm. Gözler: Işık refleksi bilateral mevcut. Pupiller izokorik. Kardiyovasküler: KTA:120/dk, S1, S2 doğal. S3 yok. Üfürüm yok, AFN +/+")
6
Fizik Muayene Solunum sistemi: Her iki hemitoraks solunuma eşit katılıyor. Toraks deformitesi yok. Retraksiyon yok. Dinlemekle ral, ronküs, ekspiryum uzunluğu yok. Gastrointestinal sistem: Batın rahat.hsm yok. Genitoüriner sistem: Haricen erkek. Anomali yok.

7
Na: mmol/L K: ,3 mmol/L Üre: mg/dL Kreatinin: 0,38 mg/dL Lökosit: /mm3 Nötrofil: /mm3 Hb: ,4 g/dL Trombosit: /mm3 Kan gazları Ter testi normal sınırlarda…

8
İshali olan hastanın gaita serolojisinde ‘adenovirus antijeni’ saptandı.
Hastada osmotik diyare düşünüldü, laktozsuz formül başlandı.

9
01/02/2012’de hasta taburcu edildi.
08/02/2012’de ateş şikayetiyle polikliniğe tekrar başvurdu.

10
Renk soluk ,cilt kuru Baş boyun: Saç ve saçlı deri doğal. Kafa yapısı simetrik. Ön fontanel 3X3 cm Gözler: Işık refleksi bilateral mevcut. Pupiller izokorik. Kardiyovasküler: KTA:120/dk, S1, S2 doğal. S3 yok. Üfürüm yok, AFN +/+ Solunum sistemi: Her iki hemitoraks solunuma eşit katılıyor. Toraks deformitesi yok. Retraksiyon yok. Dinlemekle ral, ronküs, ekspiryum uzunluğu yok.

11
Gastrointestinal sistem: Batın bombe. Hepatomegali 2cm, splenomegali yok. Traube? Ateşinin ve yüksek CRP değerinin olması nedeniyle Çocuk İnfeksiyon Hastalıkları’na (hastane kaynaklı enfeksiyon ?) danışıldı. Genel durumu iyi, toksik görünüm yok… Haliyle takibi uygun, genel durumu bozulursa antibiyotik tedavisi başlanabilir.
 danışıldı. Genel durumu iyi, toksik görünüm yok… Haliyle takibi uygun, genel durumu bozulursa antibiyotik tedavisi başlanabilir.")
12
Lökosit: /mm3 Nötrofil: /mm3 Hb: ,06 g/dL Trombosit: /mm3

13
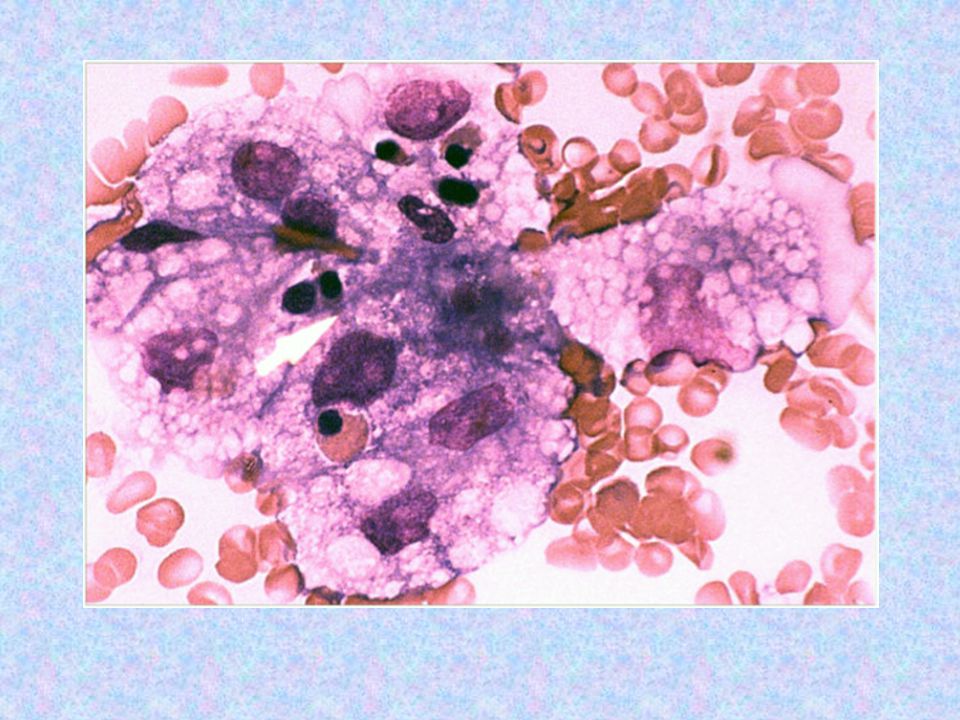
Trombositopenisi ve anemisi olan hasta Çocuk Hematoloji’ye danışıldı. PY’da lenfosit hakimiyeti mevcut, atipik hc görülmedi. Nötrofillerde toksik granülasyon, vakuolizasyon mevcut. İri trombositler izlendi. Enfeksiyonla ilişkili trombositopeni + Fizyolojik anemi Kan sayımı takibi ve tekrar danışılması uygundur.

14
12.02.2012 PT: 38,2 sn Hb: 6,48 g/dL aPTT: 61 sn Lökosit: 11900/mm3
INR: 4, MNS: /mm3 fibrinojen: mmax (düşük!!) Trombosit:16100/mm3 - K vit 1 X 3mg - TDP transfüzyonu - ES ve TS transfüzyonu
 Trombosit:16100/mm3. - K vit 1 X 3mg. - TDP transfüzyonu. - ES ve TS transfüzyonu.")
15
Hb Lökosit MNS Tromb. PT aPTT INR 12.02 6,4 11900 1880 16100 38,2 61 4,59 ES,TS, TDP 13.02 10,5 6400 1490 26700 76,9 94 9,85 TDP, TS 14.02 8,8 2850 17400 KİA 15.02 16.02 7,7 1370 875 20100 25,1 40,4 2,32 TDP 17.02 7,6 1030 12600 26,8 27,7 2,53 TS, TDP 18.02 7,81 1700 596 97800 13,7 25,6 1,19 IVIG 19.02 6,84 1100 187 51800 20.02 9,62 293 41 30600 21.02 9,58 1310 47 27400 15 31,4 1,2 TS 22.02 9,29 931 31 129000

16
Hepatomegali: 6,5 cm Splenomegali: 5,5 cm Trigliserid: 175 mg/dL HDL kolesterol: 5 LDL kolesterol: 92 VDL kolesterol: 35 Ferritin: mg/dL

17
Antı-HIV negatif CMV IgM (-), IgG(+) Rubella IgM (-), IgG(+) EBV serolojisi bekleniyor
, IgG(+) Rubella IgM (-), IgG(+) EBV serolojisi bekleniyor")
19
Hemofagositik Lenfohistiositozis

20
HLH Kalıtsal veya kazanılmış bağışıklık yetersizliği zemininde gelişen kontrolsüz aşırı yangı, Ana belirtiler: uzun süreli ateş hepatosplenomegali sitopeniler MSS semptomları sıktır.

21
HLH Biyokimyasal belirteçler:
trigliserid-ferritin seviyelerinde artış, fibrinojen seviyesinde düşüş, sIL-2 reseptörünün α zincirinde artış, NK hücreleri ve sitotoksik T hücreleri’nin fonksiyonlarında bozukluk

22
HLH Genetik geçişli tip
*Ailesel tip (perforin, munc 13-4, syntaxin 11 ve diğer bilinmeyen) *İmmun yetersizlikler _ Gricelli Sendromu _ Chediak-Higashi _ X-linked LPS
 *İmmun yetersizlikler _ Gricelli Sendromu. _ Chediak-Higashi. _ X-linked LPS.")
23
HLH Kazanılmış tip: - İnfeksiyon-ilişkili hemofagositik sendrom (IAHS)
*Eksojen Ajanlar (infeksiyonlar, toksinler) *Endojen Ürünler (doku yıkımları, metabolik ürünler) - Doğumsal metabolik hastalıklar: lizinürik protein entoleransı - Romatizmal Hastalıklar (sJRA, Still hast, SLE) Makrofaj Aktivasyon Sendromu (MAS) - Malign Hastalıklar (Lösemi, lenfoma)
 *Endojen Ürünler (doku yıkımları, metabolik ürünler) - Doğumsal metabolik hastalıklar: lizinürik protein entoleransı. - Romatizmal Hastalıklar (sJRA, Still hast, SLE) Makrofaj Aktivasyon Sendromu (MAS) - Malign Hastalıklar (Lösemi, lenfoma)")
24
HLH Klinik semptomlar karakteristik olmalarına rağmen özgül değildirler. Lenfadenopati, ikter, kraniyal sinir felci-konvulsiyon gibi nörolojik semptomlar da izlenebilir. Genellikle transaminazlar ve bilirubin seviyelerinde artış görülür. Hemofagositoz: Kemik iliği ve diğer dokularda eritrosit, lökosit ve trombositlerin histiositler tarafından fagosite edilmesi, başlangıç aşamasında hastaların çoğunda gözlenemez, hastalık ilerledikçe görülme sıklığı artar.

25
HLH Organomegali gelişimi, kan tablosundaki değişiklikler, biyokimyasal değerler bizi uyarmalı ve enfeksiyon ajanına karşı anormal cevap verildiğini düşünmeliyiz..!!! Tedavi almayan hastalarda…… NÖTROPENİ Ağır mantar ve bakteriyel infeksiyon Serebral fonksiyon bozukluğu ÖLÜM

26
Genetik HLH FHLH 1/50000, otozomal resesif geçişli
% 70 yaşamın ilk yılı içinde ortaya çıkar İmmun yetersizliklere sekonder HLH sporadik ve sık olarak ortaya çıkar

27
İmmun yetersizliklere ikincil HLH
Gricelli sendromu: hipopigmentasyon, nötrofil fonk.da bozukluk Chediak-Higashi Sendromu albinism, nötrofillerde kemotaksiste azalma ve dev inkluzyon cisimcikleri (lizozomlar) X-linked LPS EBV ilişkili HLH, lenfoma ve disgamaglobulinemi
 X-linked LPS. EBV ilişkili HLH, lenfoma ve disgamaglobulinemi.")
28
Genetik HLH Ailesel olgularda moleküler tanı: (OR)
FHLH-1 9q ilişkili gen ve fonksiyonu bilinmiyor FHLH-2 Perforin (PRF 1) geni tüm ailesel olguların %20-40 FHLH-3 UNC 13 D (17q25) sitolitik granüllerden salınımla ilişkili FHLH-4 STX11(6q24) hücre içi taşımayla ilişkili (%10-20 olguda) Türk kökenli hastalarda tanımlanmış.
 geni tüm ailesel olguların % FHLH-3 UNC 13 D (17q25) sitolitik granüllerden salınımla ilişkili. FHLH-4 STX11(6q24) hücre içi taşımayla ilişkili (%10-20 olguda) Türk kökenli hastalarda tanımlanmış.")
29
Genetik HLH XLP hastalarının %60-70 inde SAP (SLAM-ilişkili protein) SH2DIA veya DSHP olarak da adlandırılır. Xq25 genindedir. T lenfosit ve NK hücre aktivasyonunda rol alır. Chediak-Higashi sendromunda lizozomlarla ilişkili LYST geni (1q42) GS-2: Griscelli sendromu tip 2 de sitotoksik granül eksositozu ve plazma membranına taşıma ile ilişkili RAB27a geni
 SH2DIA veya DSHP olarak da adlandırılır. Xq25 genindedir. T lenfosit ve NK hücre aktivasyonunda rol alır. Chediak-Higashi sendromunda lizozomlarla ilişkili LYST geni (1q42) GS-2: Griscelli sendromu tip 2 de sitotoksik granül eksositozu ve plazma membranına taşıma ile ilişkili RAB27a geni.")
30
Sekonder HLH Çocuklar ve erişkinlerdeki insidans bilinmiyor,
Ancak düşünüldüğünden çok daha sık görülmekte, Çoğunlukla viruslar, ve bakteri, mantar, protozoalarla tetiklenebilir. Bir çalışmada etkenlerin %74’ü EBV, Sadece destek tedavisiyle %50 mortalite… İnfeksiyon etkeninin saptanması FHLH ve sekonder HLH ayrımını sağlamaz.

31
Primer-Sekonder HLH ayrımı
Sekonder olgular aile öyküsü, akraba evliliği ve özellikle genetik kusurun bulunmaması ile familyal vakalardan ayrılır. Daha önce tanımlanmış bir genetik kusur saptanamıyorsa dahi hastanın yaşı küçükse, anne-baba akraba ise ve ailede benzer hastalık öyküsü varsa primer HLH akla gelmelidir. Familyal hemafagositozun da enfeksiyonun tetiklemesi ile başlayabildiği, özellikle hastalığın ağır ve uzun sürdüğü ve tekrarlamalar gösterdiği durumda familyal olabileceği hatırlanmalıdır .

32
HLH de tanı kriterleri 1.Ateş 2.Splenomegali
3.Sitopeni (Üç seriden en az ikisinde sitopeni) Hb < 9 g/dl, yenidoğan ise Hb<12 g/dl trombosit < /mm3, nötrofil < 1000/mm3 4.Hipertrigliseridemi ve/veya hipofibrinojenemi açlık Trig>265 mg/dL hipofibrinojenemi <1.5 g/L 5.NK aktivitesinin olmaması ya da azalmış olması 6.Ferritin ≥500 mikrogram/L 7.Soluble CD25≥ 2400 U/ml 8.Kemik iliği, lenf nodu ya da dalakta malinite olmaksızın hemofagositoz gözlenmesi
 Hb < 9 g/dl, yenidoğan ise Hb<12 g/dl. trombosit < /mm3, nötrofil < 1000/mm3. 4.Hipertrigliseridemi ve/veya hipofibrinojenemi. açlık Trig>265 mg/dL. hipofibrinojenemi <1.5 g/L. 5.NK aktivitesinin olmaması ya da azalmış olması. 6.Ferritin ≥500 mikrogram/L. 7.Soluble CD25≥ 2400 U/ml. 8.Kemik iliği, lenf nodu ya da dalakta malinite olmaksızın hemofagositoz gözlenmesi.")
33
8 kriterin en az 5 tanesinin olması Veya HLH’nin moleküler tanısı
Tanı için: 8 kriterin en az 5 tanesinin olması Veya HLH’nin moleküler tanısı (bu durumda tüm kriterlerin tamamlanması şart değildir) Ancak HLH bazen atipik ya da sinsi bir seyir izleyebilir. Bu durumda tüm kriterler olmayabilir. Bazı hastalarda da diğer kriterler zaman içinde tamamlanabilir.
 Ancak HLH bazen atipik ya da sinsi bir seyir izleyebilir. Bu durumda tüm kriterler olmayabilir. Bazı hastalarda da diğer kriterler zaman içinde tamamlanabilir.")
34
Tanı Güçlüğü Ayırıcı tanıya enfeksiyonların girmesi, enfeksiyon hastalıklarına yönelik ayrıntılı tetkikler ve antibiyotik kullanımı ile vakit kaybına neden olmaktadır. Saptanan bir enfeksiyon etkeni ile tüm tablo açıklanmaya çalışılmaktadır. Genellikle organomegalili bebeklerde metabolik taramalar başlatılır. Çoğu zaman sepsis tanısı konur ancak tablonun ağırlığı bir immün sistem kusurunu akla getirmelidir.

35
Tedavinin Amaçları İnflamasyonu baskılamak
Patojenle enfekte hücreleri yok etmek Enfeksiyon etkenine yönelik tedavi (Leishmania ilişkili HLH dışında genellikle tek başına yeterli olmaz) Genetik geçişli olgularda HSCT ile kusurlu immün sistemi düzeltmek
 Genetik geçişli olgularda HSCT ile kusurlu immün sistemi düzeltmek.")
36
Hasta Yaş Yıl Akr. İlişkili Hast. Genetik/ Edinsel Mutasyon Nörolojik tutulum Prognoz 1 2 ay (-) Genetik ? Yaşıyor +8 ay, başka merkezde 2 8,1 (+) Enfeksiyon İyileşme, +12 ay 3 7,5 CHS 2.remisyon, +15 ay 4 9,2 EBV İyileşme, 5 7 ay Ailesel Munc 13-4 exon 23 Eksitus, 1 ay 6 1,7 JİA Perforin (-) Syntaxin (-) Munc 13-4 (-) İyileşme, +15 ay 7 3,5 ay + Eksitus, 5 ay (reaktivasyon) 8 11 y Perforin val50 Thr Eksitus, 48 ay, (3 reaktivasyon) 9 1 gün Eksitus, 13 gün 10 13 c.1349C>T, p.T450M homozigot perforin mutasyonu Yaşıyor 2. kısmi remisyon, 5,5 ay 11 7,5 ay Griscelli sendr. RAB27 (A148 C) 1-bp delesyonu (del G) + Reaktivasyon sırasında Exitus 5 ay 12 Leishmaniasis - Şifa, yaşıyor, 9 ay 6 ay +3ay remisyonda yaşıyor CHS: Chediak Higashi-Sendromu, EBV : Ebstein Barr Virus , JİA: Juvenil idyopatik artrit
 Genetik. Yaşıyor +8 ay, başka merkezde. 2. 8,1. (+) Enfeksiyon. İyileşme, +12 ay. 3. 7,5. CHS. 2.remisyon, +15 ay. 4. 9,2. EBV. İyileşme, 5. 7 ay. Ailesel. Munc 13-4 exon 23. Eksitus, 1 ay. 6. 1,7. JİA. Perforin (-) Syntaxin (-) Munc 13-4 (-) İyileşme, +15 ay. 7. 3,5. ay. + Eksitus, 5 ay. (reaktivasyon) y. Perforin val50 Thr. Eksitus, 48 ay, (3 reaktivasyon) 9. 1 gün. Eksitus, 13 gün c.1349C>T, p.T450M homozigot perforin mutasyonu. Yaşıyor 2. kısmi remisyon, 5,5 ay ,5 ay. Griscelli sendr. RAB27 (A148 C) 1-bp delesyonu (del G) + Reaktivasyon sırasında. Exitus 5 ay. 12. Leishmaniasis. - Şifa, yaşıyor, 9 ay. 6 ay. +3ay remisyonda yaşıyor. CHS: Chediak Higashi-Sendromu, EBV : Ebstein Barr Virus , JİA: Juvenil idyopatik artrit.")
37
Tedavi

38
Tedavi Kemi iliği transplantasyonu
















>")




