Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Doğuştan karbohidrat metabolizması bozuklukları

3
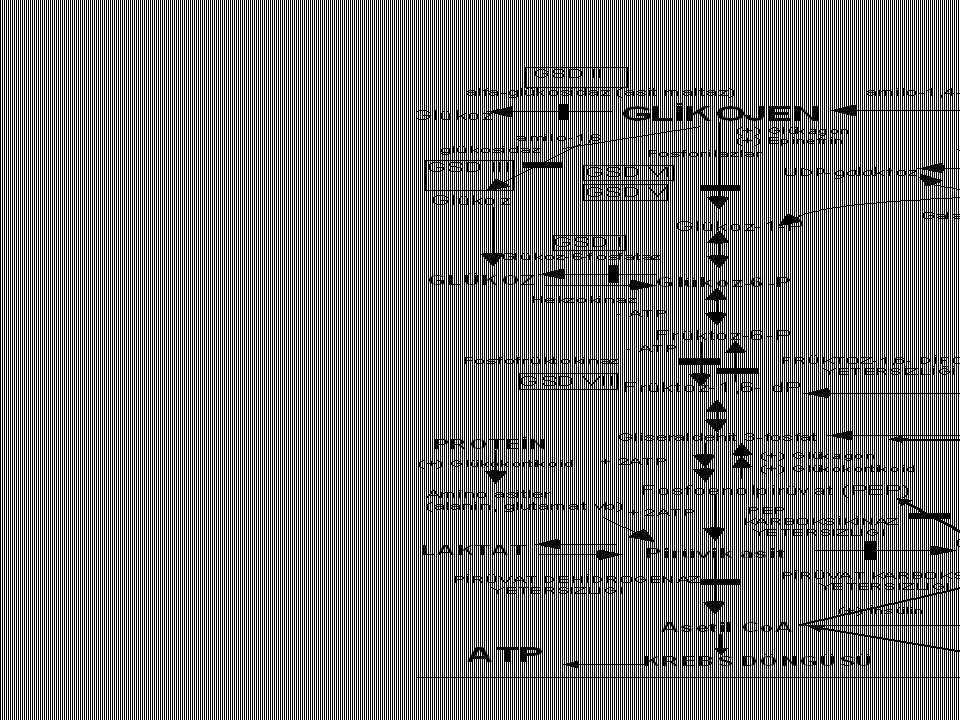
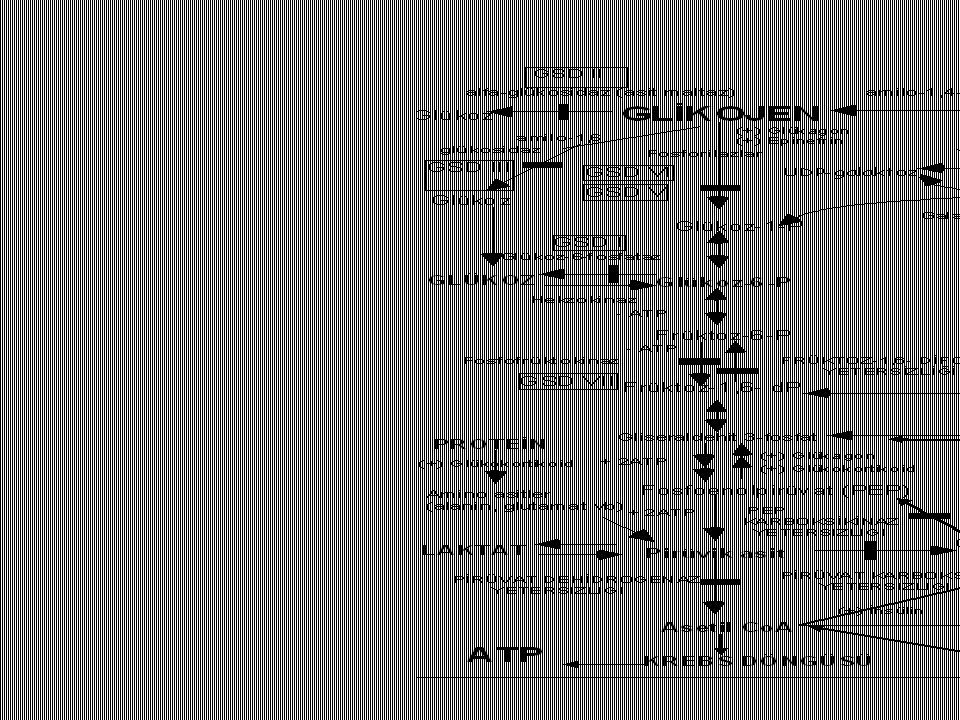
KLASİK GALAKTOZEMİ Defektif enzim: Galaktoz-1-fosfat üridiltransferaz (1:40,000) Mekanizma: Galaktoz-1-fosfat birikimi hücre içi ATP deplesyonuna neden olur (fosforu bağlar); ayrıca karaciğer, böbrek ve beyin için çok toksiktir.
; ayrıca karaciğer, böbrek ve beyin için çok toksiktir.")
4
Klasik galaktozemi Galaktoz Galaktoz-1P Glükoz Galaktitol galaktokinaz
Gal 1-P üridil transferaz epimeraz Glükoz

5
Galaktozemi-Klinik/laboratuar
İshal Kusma Dehidratasyon Tartı kaybı Hepatomegali Noktasal kataraktlar (galaktitol artışı) KC yağlı dejenerasyon ve siroz Sepsis (E. Coli) Direkt hiperbilirubinemi Renal tübüler hasar PT ve aPTT’de uzama Transaminazlarda yükselme
 KC yağlı dejenerasyon ve siroz. Sepsis (E. Coli) Direkt hiperbilirubinemi. Renal tübüler hasar. PT ve aPTT’de uzama. Transaminazlarda yükselme.")
6
Galaktozemi-Tanı İdrarda glükoz ve galaktoz artışına bağlı indirgeyici madde varlığı (Fehling veya Benedict testi) Eritrositlerde galaktoz-1-fosfat üridiltransferaz aktivitesinin düşüklüğü (Beutler testi). Serumda ve eritrositlerde galaktoz ve galaktoz-1-fosfat (N: mg/dL) yüksekliği. Renal tübüler hasar: Generalize aminoasidüri, albüminüri (Fanconi sendromu)
. Serumda ve eritrositlerde galaktoz ve galaktoz-1-fosfat (N: mg/dL) yüksekliği. Renal tübüler hasar: Generalize aminoasidüri, albüminüri (Fanconi sendromu)")
7
Galaktozemi-Tedavi- Komplikasyonlar
Tedavi: Ömür boyu laktozsuz diyet Komplikasyonlar - Tedaviye rağmen hafif mental retardasyon - Ataksi, tremor - Gonadal bozukluklar/ovaryum kistleri

8
Epimeraz yetersizliği
Galaktoz Galaktitol galaktokinaz Galaktoz-1P Gal 1-P üridil transferaz epimeraz Glükoz

9
Epimeraz yetersizliği
Hafif formasında sadece eritrositlerde galaktoz-1-fosfat birikimi vardır, fakat hiçbir klinik belirti görülmez. Ağır formasının klinik ve biyokimyasal bulguları klasik galaktozemiye benzer.

10
Galaktokinaz yetersizliği
Galaktoz Galaktitol galaktokinaz Galaktoz-1P Gal 1-P üridil transferaz epimeraz Glükoz

11
Galaktokinaz yetersizliği
Galaktoz ve galaktitol birikimi hızla ilerleyen bilateral santral katarakt katarakta neden olur. Galaktoz toksisitesine ait diğer belirtiler yoktur. Tedavide laktozsuz diyet uygulanır.

12
Kalıtsal früktoz entoleransı
Defektif enzim: Aldolaz B (OR, 1:20,000) Mekanizma: Früktoz-1-fosfat birikimi hücre içi ATP deplesyonuna neden olur (fosforu bağlar); ayrıca karaciğer, böbrek ve beyin için çok toksiktir.
 Mekanizma: Früktoz-1-fosfat birikimi hücre içi ATP deplesyonuna neden olur (fosforu bağlar); ayrıca karaciğer, böbrek ve beyin için çok toksiktir.")
13
Kalıtsal Früktoz entoleranı (Früktozemi)
Früktokinaz Früktoz-1P Aldolaz Glükoz

14
Früktozemi-Klinik Kusma, apati , kilo kaybı,
Ağır karaciğer fonksiyon bozuklukları, hepatomegali Hipoglisemi Renal tubüler bozukluklar Früktozlu gıdalara karşı tiksinti Diş çürüğü yok (!!).
.")
15
Früktozemi-Tanı Früktoz yükleme sırasında bulguların ortaya çıkması (tanısal fakat tehlikeli !!!), Diyet ile bulguların düzelmesi, enzim çalışması. Renal tübüler hasar: Generalize aminoasidüri, albüminüri İdrarda glükoz ve galaktoz artışına bağlı indirgeyici madde varlığı (Fehling veya Benedict testi) Tedavi: Hayat boyu früktozsuz diyet.
 Tedavi: Hayat boyu früktozsuz diyet.")
16
Esansiyel früktozüri Früktoz Früktokinaz Früktoz-1P Aldolaz Glükoz

17
Esansiyel früktozüri Defektif enzim: Früktokinaz Klinik bulgular: Yok.
Tanı: İdrarda glükoz dışı redüktan madde (Teşhis tesadüfe bağlı)
")
18
Glikojen depo hastalıkları

20
Glikojen depo hastalıkları- Sınıflama
Daha çok karaciğeri tutan ve hipoglisemiye neden olanlar (tip 0, I, VI) Daha çok kasları tutan ve aneorobik metabolizmayı bozanlar (Tip II, V, VII) Hem karaciğer, hem kasları tutan ve hipoglisemi yapan (Tip III). Karaciğer sirozu yapanlar: (Tip III , Tip IV).
 Daha çok kasları tutan ve aneorobik metabolizmayı bozanlar (Tip II, V, VII) Hem karaciğer, hem kasları tutan ve hipoglisemi yapan (Tip III). Karaciğer sirozu yapanlar: (Tip III , Tip IV).")
21
GSD tip O Karaciğer glikojen sentaz eksikliği
Sabah erken saatlerde konvülziyonlar tipik Glukagona yanıtsız hipoglisemi Hepatomegali yok

22
Tip I a (von Gierke)/ Klinik/Laboratuar
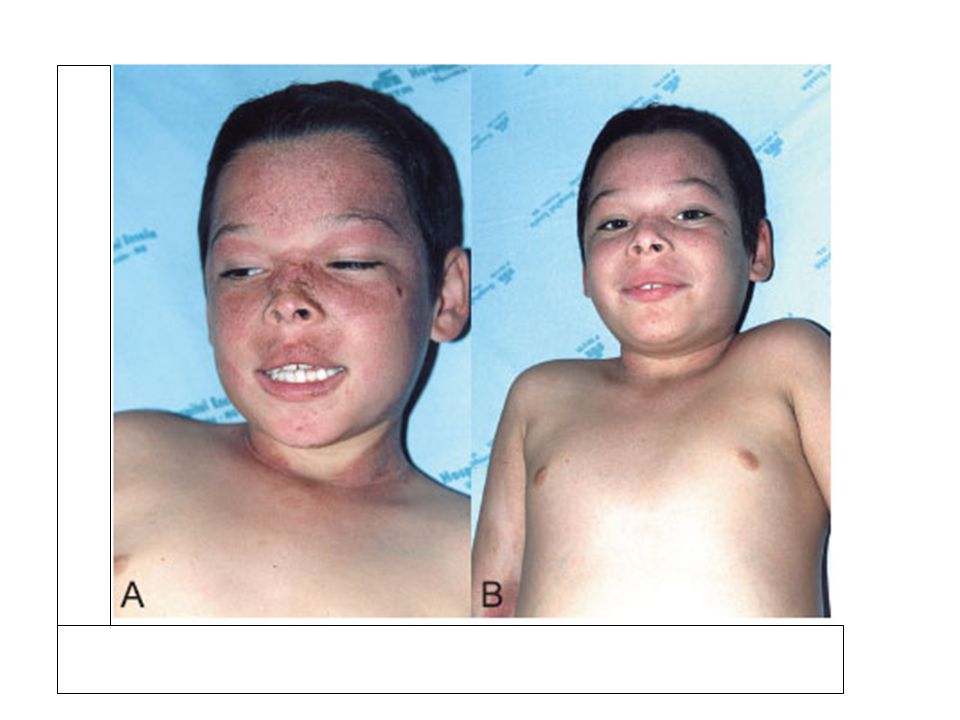
Neonatal hipoglisemi Laktik asidoz, ketozis Hepatomegali Hiperürisemi gut Lipoliz hiperlipidemiye yol açar. Yanaklarda lipid birikimi olur (taş bebek yüzü).
.")
23
Tip I a (von Gierke)/Tanı
Hastalara glükagon verildiğinde normalde beklenen hiperglisemi genellikle saptanamaz. Glükoz yüklemesinden sonra glükoz yükselirken laktat düzeyi düşer. Kesin tanı karaciğerde glükoz-6-fosfataz düşüklüğünün gösterilmesi

24
Tip I a (von Gierke)/Tedavi
Tedavide temel ilke glükoneogenezin mümkün olduğunca azaltılmasıdır. Geceleri yapılan enteral beslenme idealdir, fakat pratik değildir. Öğünler sıklaştırılır ve hastalara bağırsaktan yavaş emilen pişmemiş nişasta (mısır, pirinç) gibi glükoz polimerileri verilir. Mümkün olduğunca süt ürünleri diyetten çıkarılır.
 gibi glükoz polimerileri verilir. Mümkün olduğunca süt ürünleri diyetten çıkarılır.")
25
GSD tip Ib Klinik GSD tip Ia gibidir.
Ek olarak nötropeni, tekrarlayan enfeksiyonlar ve Crohn benzeri enflamatuvar barsak hastalığı vardır. Splenomegali olabilir. Glukoz 6 fosfatın mikrozomal membrandan transportunda bozukluk vardır (mikrozomal transport protein T1 bozukluğu) Nötropeni için G-CSF ve GM-CSF kullanılır.
 Nötropeni için G-CSF ve GM-CSF kullanılır.")
26
GSD tip Ic ve Id Klinik GSD tip Ia gibidir
Ic: T2 protein bozukluğuna bağlı inorganik fosfat transportu bozuk Id: GLUT 7 mikrozomal transport bozukluğu

27
GSD tip VI Hers hastalığı
Karaciğer fosforilaz eksikliği Büyüme geriliği ve hepatomegali Selim gidişli Yaşla hepatomegali geriler Hipoglisemi

28
GSD tip VII Tarui hastalığı
Kas, eritrosit ve trombositlerde fosfofruktokinaz eksikliği Tip V’ten daha ağır miyopati Hafif hemolitik anemi Egzersizle artan hiperürisemi PAS pozitif anormal amilopektin birikimi Yemek sonrası egzersiz entoleransı (Tip V’ten farklı olarak glukoz infüzyonuna cevapsız)
")
29
GSD tip IX Fosforilaz kinaz eksikliği
X’e bağlı karaciğer formu (en sık) OR karaciğer ve kas formu OR karaciğer formu Kas formu Kalp formu
 OR karaciğer ve kas formu. OR karaciğer formu. Kas formu. Kalp formu.")
30
GSD tip XI (Fanconi-Bickel sendromu)
GLUT 2 eksikliği GSD Ia benzeri bulgular Boy kısalığı, Fanconi sendromu, raşitizm Açlık hipoglisemisi, tokluk hiperglisemisi Karaciğer ve böbrekte glikojen birikimi

31
GSD tip II (Pompe hastalığı)
Enzim: alfa-1,4-glükosidaz (asit maltaz) bir lizozomal enzimdir; glükoz hemeostazındaki rolü önemsizdir. İnfantil, çocukluk ve erişkin olmak üzere üç tipi vardır. İnfantil tip en ağır forma olup yenidoğan döneminden itibaren belirti vermeye başlar. Ağır bir hipotoni mevcuttur. Karaciğer büyüktür. Kardiyomegali erken bir dönemde gelişir. Lenfositler vakuollüdür. EKG'de bütün derivasyonlarda PR-kısalığı ve QRS yükseklikleri mevcuttur. Kalp yetersizliği genellikle ilk altı ay içerisinde ölümle sonuçlanır. Enzim tedavi başarılıdır.
 bir lizozomal enzimdir; glükoz hemeostazındaki rolü önemsizdir. İnfantil, çocukluk ve erişkin olmak üzere üç tipi vardır. İnfantil tip en ağır forma olup yenidoğan döneminden itibaren belirti vermeye başlar. Ağır bir hipotoni mevcuttur. Karaciğer büyüktür. Kardiyomegali erken bir dönemde gelişir. Lenfositler vakuollüdür. EKG de bütün derivasyonlarda PR-kısalığı ve QRS yükseklikleri mevcuttur. Kalp yetersizliği genellikle ilk altı ay içerisinde ölümle sonuçlanır. Enzim tedavi başarılıdır.")
32
GSDTip III (Cori / Forbes hastalığı)
Cori hastalığında amilo-1, 6-glükosidaz eksikliği nedeni ile karaciğer ve kas dokusunda sınır dekstrini birikimi mevcuttur. Hastalık genellikle süt çocuğu döneminde, açlık hipoglisemisi, hepatomegali ve büyüme geriliği ile karşımıza çıkar. Bazı olgularda kalp ve kas tutulması da görülebilir. Laboratuar bulguları Tip I gibi fakat ondan daha hafiftir. Tedavide hasta sık beslenir.

33
GSD Tip IV (Andersen hastalığı)
Dallandırıcı enzim eksikliğine bağlıdır. Karaciğer biyopsilerinde amilopektine benzer anormal yapıda bir glikojen molekülü bulunur. Bu molekülün suda erimesi az olduğundan, yabancı cisim gibi reaksiyon vererek karaciğer sirozuna ve erken çocukluk çağında ölüme neden olur.

34
GSD Tip V (McArdle hastalığı)
Kas fosforilazı eksiktir. Hipoglisemiye neden olmaz. Adolesan ve erişkinlik döneminde bulgu verir. Başlıca bulgular, efora tahammülsüzlük, kramplar ve miyoglobinüridir. İskemi sırasında (örneğin tansiyon aletinin manşonu ile kol sıkıldığında) normalde beklenen laktik asit yükselmesi gerçekleşmez.
 normalde beklenen laktik asit yükselmesi gerçekleşmez.")
35
GLUT1 yetersizliği Epilepsi, mikrosefali, motor mental gerilik ile karakterizedir. BOS glükozu kan glükozunun %35’inden daha azdır (normalde % 65 kadardır). BOS laktatı düşük olabilir. Erken dönemde ketojenik diyet (düşük şekerli, yüksek yağlı) yapılırsa prognoz iyidir.
. BOS laktatı düşük olabilir. Erken dönemde ketojenik diyet (düşük şekerli, yüksek yağlı) yapılırsa prognoz iyidir.")
36
GLUT2 yetersizliği (Fanconi-Bickel sendromu)
Monosakkaritlerin tübüler geri emilimi ve barsaktan monosakkarit emilimi bozuktur. Klinikte boy kısalığı, raşitizm ve hepatomegali ile karşımıza çıkar; böbrekler büyüktür. OGTT’de açlık hipoglisemisi ve tokluk hiperglisemisi vardır. Ayrıca barsak emilimi ve böbrek geri emilimi bozulduğundan idrar ve gaitada monosakkarit saptanır. Genaralize hiperaminoasidüri, hiperfosfatüri, hiperürikozüri ve hiperkalsüri mevcuttur. .

37
Kongenital glükoz-galaktoz malabsorpsiyonu
Hastalık SGLT1 (sodyumla eşleşmiş glukoz taşıyıcısı) yetersizliğine bağlıdır. İntestinal glükoz ve galaktoz transportunun ağır derecede bozulduğu nadir bir defekttir. Renal transporttaki kusur ise hafiftir. Hastalık yenidoğan döneminde sulu ve asidik ishallerin yol açtığı ağır dehidratasyon atakları ile karakterizedir. Sabit ya da intermitan glükozüri sıktır. Dışkıda redüktan madde pozitif olup nefesteki hidrojen miktarı artmıştır. Glükoz ve galaktozsuz diyet (fruktozun emilimi bozulmamıştır) ile dramatik iyileşme olur.
 yetersizliğine bağlıdır. İntestinal glükoz ve galaktoz transportunun ağır derecede bozulduğu nadir bir defekttir. Renal transporttaki kusur ise hafiftir. Hastalık yenidoğan döneminde sulu ve asidik ishallerin yol açtığı ağır dehidratasyon atakları ile karakterizedir. Sabit ya da intermitan glükozüri sıktır. Dışkıda redüktan madde pozitif olup nefesteki hidrojen miktarı artmıştır. Glükoz ve galaktozsuz diyet (fruktozun emilimi bozulmamıştır) ile dramatik iyileşme olur.")
38
Renal glükozüri Renal glükozüri, glükozun renal transportunu bozan otozomal resessif bir hastalıktır; SGLT2 yetersizliğine bağlıdır. İntestinal glükoz ve galaktoz transportunda defekt yoktur. Tip A'da renal glükozüri eşiği ve maksimal tubüler reabsorpsiyon azalmıştır. Tip B'de renal glükozüri eşiği azalmış fakat maksimal tubüler reabsorpsiyon normaldir. Tip O'da tubüler reabsorpsiyon hiç yoktur. İzole renal glükozüri nadiren semptom verir. Rutin idrar tahlilleri sırasında tesadüfî olarak tanısı konulur.

39
Sistinüri Defekt: Dibazik amino asitlerin (lizin, arjinin, ornitin, ve sistin) renal tübüler reabsorpsiyonunda azalma Klinik bulgular: Nefrolitiazis (sistin 1250 µmol/L’nin üzerinde ve asit pH’da kristalleşir) Tanı: Taze idrarda nitroprussid testi pozitiftir. İdrarda lizin, arjinin, ornitin, ve sistin boşaltımı artmıştır. Tedavi: Fazla (>5L) sıvı alma, idrarın alkalileştirilmesi (üriner enfeksiyonlar !). Seçilmiş olgularda penisillamin (1-2 g/ gün), merkaptopropionilglisin veya kaptopril.
 Tanı: Taze idrarda nitroprussid testi pozitiftir. İdrarda lizin, arjinin, ornitin, ve sistin boşaltımı artmıştır. Tedavi: Fazla (>5L) sıvı alma, idrarın alkalileştirilmesi (üriner enfeksiyonlar !). Seçilmiş olgularda penisillamin (1-2 g/ gün), merkaptopropionilglisin veya kaptopril.")
40
Hartnup hastalığı Defekt: Nötral amino asitlerin (alanin, valin, treonin, lösin, izolösin, fenilalanin, tirozin, triptofan, histidin, glisin) intestinal ve renal tübüler reabsorpsiyonunda azalma; triptofan yetersizliği niasin (pellegra) ve serotonin yetersizliğine yol açar. Klinik bulgular: Fotodermatit, serebellar ataksi; çoğu kez asemptomatik Tanı: İdrarda nötral amino asitlerin yüksekliği, plazmada düşüklüğü. Tedavi: Niasin mg/gün, güneşten korunma, proteinden zengin diyet
 intestinal ve renal tübüler reabsorpsiyonunda azalma; triptofan yetersizliği niasin (pellegra) ve serotonin yetersizliğine yol açar. Klinik bulgular: Fotodermatit, serebellar ataksi; çoğu kez asemptomatik. Tanı: İdrarda nötral amino asitlerin yüksekliği, plazmada düşüklüğü. Tedavi: Niasin mg/gün, güneşten korunma, proteinden zengin diyet.")
42
Metionin Malabsorbsiyonu (şerbetçiotu kokulu idrar hastalığı)
Böbrek tübülleri ve incebağırsakta metionin emilimi bozulmuştur. Klinik bulgular: saçların beyaz olması, konvülsiyon nöbetleri, sürgün, ödem, zeka geriliği ve idrar kokusu (biraya benzer). Tedavi: metioninden fakir diyet.
. Tedavi: metioninden fakir diyet.")
43
Kistik Fibroz

44
Kistik fibroz/Etyoloji/ Epidemiyoloji
Kistik fibroz ekzokrin bezlerin kronik ve ilerleyici yetersizliği ile karakterize otozomal resesif geçişli bir hastalıktır. Kistik fibroz geni (7q 31.2) kistik fibroz transmembran iletisi düzenleyen proteini (CFTR) kodlar. CFTR, cAMP ile uyarılan klor kanallarının proteinidir. Kistik fibroz geninin 1500’den fazla mütasyonu bildirilmiştir. Bunlardan en sık (Dünyada%70, Türkiyede %20) görüleni delta 508 mutasyonudur. Beyaz ırktaki sıklığı 1:2000-1:3000 arasındadır.
 kistik fibroz transmembran iletisi düzenleyen proteini (CFTR) kodlar. CFTR, cAMP ile uyarılan klor kanallarının proteinidir. Kistik fibroz geninin 1500’den fazla mütasyonu bildirilmiştir. Bunlardan en sık (Dünyada%70, Türkiyede %20) görüleni delta 508 mutasyonudur. Beyaz ırktaki sıklığı 1:2000-1:3000 arasındadır.")
45
Kistik fibrozda solunum epitelinde görülen sıvı sekresyonu ve emilimindeki bozukluklar

46
Ter sekresyonu

47
Kistik fibroz: Patogenez

48
Kistik fibrozda başvuru bulguları
Başvuru bulgusu % Akut ya da persistan solunum semptomları Büyüme gelişme geriliği ve malnütrisyon Anormal dışkı Mekonyum ileusu, barsak obstrüksiyonu Aile hikayesi Psödo-Bartter sendromu Rektal prolapsus Nazal polip, sinüzit Hepatobiliyer hastalık Diğer 50 43 35 19 17 5 3 2 1 <1

49
Kistik Fibrozda Klinik Bulgular- Alt solunum yolu
Reaktif hava yolu hastalığı Tekrarlayan bronşiolit Bronşektazi (üst loblar), çomak parmak Tekrarlayan pnömoni Atelektazi, pnömotoraks Hemoptizi Allerjik bronkopulmoner aspergilloz Solunum yetersizliği Kor pulmonale
, çomak parmak. Tekrarlayan pnömoni. Atelektazi, pnömotoraks. Hemoptizi. Allerjik bronkopulmoner aspergilloz. Solunum yetersizliği. Kor pulmonale.")
50
Kistik fibroz: KOAH

51
Balgam ya da boğaz kültüründe en çok kolonize olan bakteriler
S.aureus (ilk üreyen) Tiplendirilemeyen H.influenzae, P.aeruginosa (non-mukoid ve mukoid; en sık kolonize olan), Stenotrophomonas maltophilia, Achromobacter xylooxidans, Burkholderia cepacia (prognozu en çok kötüleştiren). Atipik mikobakteri (en sık M.avium, ikinci M.abscessus) Aspergillus fumigatus
 Tiplendirilemeyen H.influenzae, P.aeruginosa (non-mukoid ve mukoid; en sık kolonize olan), Stenotrophomonas maltophilia, Achromobacter xylooxidans, Burkholderia cepacia (prognozu en çok kötüleştiren). Atipik mikobakteri (en sık M.avium, ikinci M.abscessus) Aspergillus fumigatus.")
52
Kistik Fibrozda Klinik Bulgular- GİS
Mekonyum ileusu (yenidoğanda) Distal intestinal obstrüksiyon (sütçocuğu) Rektal prolapsus İnvajinasyon Kronik yağlı ishal GÖR (asit ya da safra reflüsü)
 Distal intestinal obstrüksiyon (sütçocuğu) Rektal prolapsus. İnvajinasyon. Kronik yağlı ishal. GÖR (asit ya da safra reflüsü)")
53
Mekonyum ileusu Sekresyonların koyulaşması yenidoğan döneminde mekonyum ileusuna (%10-20), daha büyük yaşlarda ise distal intestinal obstrüksiyona (mekonyum ileusu eşdeğeri) yol açabilir. Mekonyum ileuslu hastaların direkt karın grafisinde sağ alt kadranda, mekonyum içinde hapsolmuş hava kabarcıkları nedeni ile granüler bir manzara görülür. Fizik muayenede halat gibi kitle ele gelebilir. Baryumlu lavmanlar ile obstrüksiyon redükte edilemezse, mekonyum kitlesi ameliyat ile çıkartılır.
, daha büyük yaşlarda ise distal intestinal obstrüksiyona (mekonyum ileusu eşdeğeri) yol açabilir. Mekonyum ileuslu hastaların direkt karın grafisinde sağ alt kadranda, mekonyum içinde hapsolmuş hava kabarcıkları nedeni ile granüler bir manzara görülür. Fizik muayenede halat gibi kitle ele gelebilir. Baryumlu lavmanlar ile obstrüksiyon redükte edilemezse, mekonyum kitlesi ameliyat ile çıkartılır.")
54
Mekonyum ileusu

55
Kistik Fibrozda Klinik Bulgular
Ekzokrin yetersizlik (malabsorpsiyon, hipoproteinemi) Tekrarlayan pankreatit İnsülin gerektiren Diabetes Mellitus (>10 yaş) Hepatobiliyer Uzamış yenidoğan sarılığı Neonatal kolestaz Bilier siroz (fokal) Portal hipertansiyon Hepatosplenomagali Kolesistit ve kolelityazis (>20 yaş)
 Tekrarlayan pankreatit. İnsülin gerektiren Diabetes Mellitus (>10 yaş) Hepatobiliyer. Uzamış yenidoğan sarılığı. Neonatal kolestaz. Bilier siroz (fokal) Portal hipertansiyon. Hepatosplenomagali. Kolesistit ve kolelityazis (>20 yaş)")
56
Kistik Fibrozda Klinik Bulgular
Üst solunum yolu Nazal polipozis Pansinüzit Diğer Hipokloremik hipopotasemik hiponatremik alkaloz. Puberte gecikmesi İnguinal herni, inmemiş testis riskinde artış Azospermi (Epididim, vas deferens atrezisi) Endoservisitler nedeni ile doğurganlıkta azalma
 Endoservisitler nedeni ile doğurganlıkta azalma.")
57
Yalancı-Bartter sendromu
Çevre ısısı artar ve/veya diyetteki tuz azalırsa, kistik fibrozlu hastalarda tuz kaybı ve dehidratasyon gelişebilir. Tuz kaybını kompanse etmek için artan aldosteron hipopotasemi ve metabolik alkaloza (yalancı-Bartter sendromu) yol açar; idrar sodyumu düşüktür. Gerçek Bartter sendromunda idrar sodyumu yüksektir (>30mEq/L).
 yol açar; idrar sodyumu düşüktür. Gerçek Bartter sendromunda idrar sodyumu yüksektir (>30mEq/L).")
58
Kistik Fibroz-Tanı Kistik fibroza (KF) ait bir veya birden fazla fenotipik özelliğin olması veya Kardeşte KF öyküsü veya Pozitif yenidoğan tarama testi (kanda immunoreaktif tripsinojen yüksekliği) + Ter testi > 60 mEq/L (en az iki kez) veya CFTR genlerinde KF’a neden olan mutasyonların gösterilmesi
 + Ter testi > 60 mEq/L (en az iki kez) veya. CFTR genlerinde KF’a neden olan mutasyonların gösterilmesi.")
59
Ter testi Olguların az bir bölümünde terdeki sodyum ya da klorür mEq/L arasında ya da normal sınırlar içinde olabilir. Bu nedenle kuşkulu hastalarda DNA araştırması yapılması gerekir. Teknik hata, hipoproteinemik ödem ve nadir bazı mutasyon durumunda ( Kb C>T; R117H; G551S) yalancı negatif sonuçlar da görülebilir. Şüphe halinde test negatif çıksa bile tekrarlanmalıdır.
 yalancı negatif sonuçlar da görülebilir. Şüphe halinde test negatif çıksa bile tekrarlanmalıdır.")
60
Ter testinde yalancı pozitiflik yapan başlıca hastalıklar
Adrenal yetersizlik Hipotiroidi Hipoparatiroidi Nefrojen d. insipitus Psödohipoaldosteronizm Glikojen depo hastalığı I Mukopolisakaridozlar Fukosidoz Anoreksia nervosa, Pankreatit Malnütrisyon (en sık) G6PD eksikliği Familiyal kolestaz Ektodermal displazi
 G6PD eksikliği. Familiyal kolestaz. Ektodermal displazi.")
61
Neonatal tarama Pankreas kanallarının tıkanmasına bağlı olarak bağırsağa atılamaması nedeni ile kistik fibrozlu yenidoğanlarda serum tripsinojen düzeyi (immünoreaktif tripsin) artar. 3 aydan sonra pankreas tahribatı nedeni ile serum tripsinojen düzeyi düştüğünden bu test artık tarama için kullanılamaz. Erken tanı koyma hastalığın prognozunda belirgin bir iyilik sağlanmadığı için birçok gelişmiş ülkede kistik fibroz tarama programları yüksek maaliyeti nedeni ile terk edilmiştir.
 artar. 3 aydan sonra pankreas tahribatı nedeni ile serum tripsinojen düzeyi düştüğünden bu test artık tarama için kullanılamaz. Erken tanı koyma hastalığın prognozunda belirgin bir iyilik sağlanmadığı için birçok gelişmiş ülkede kistik fibroz tarama programları yüksek maaliyeti nedeni ile terk edilmiştir.")
62
Solunum yolu enfeksiyonlarının tedavisi
Postüral drenaj ve göğüs masajı ile mukosiliyer klirens arttırılır. Mukolitik ve ekspektoran şuruplar da mukus klirensini arttırmada başarılı değillerdir. Aerosol DNaz tedavisi ile DNA'yı parçalayarak mukusu sulandırmak mümkündür, fakat bu tedavi çok pahalıdır. Amilorid kullanımı artan su reabsorpsiyonunu engelleyerek mukusun koyulaşmasını azaltabilir. Zerdeçal ve hipertonik NaCl (%3-%7) inhalasyonu balgamı sulandıran daha ucuz ve etkin seçeneklerdir.
 inhalasyonu balgamı sulandıran daha ucuz ve etkin seçeneklerdir.")
63
Diyet Kalori miktarı enfeksiyonsuz hastada % , enfeksiyonlu hasta ise % oranında arttırılmalıdır. Malabsorpsiyonun azaltılması için mide asidine dayanıklı, bağırsakta mikrokürecikler şeklinde çözünen pankreas preparatları (Kreon®) kullanılır. Yağda eriyen vitaminlerin ( A, E, D, K) miktarı steatore ile orantılı olarak arttırılmalıdır. Nadiren çinko ve selenyum yetersizliği gelişebilir.
 kullanılır. Yağda eriyen vitaminlerin ( A, E, D, K) miktarı steatore ile orantılı olarak arttırılmalıdır. Nadiren çinko ve selenyum yetersizliği gelişebilir.")
Benzer bir sunumlar





>")













