Sunuyu indir
1
Uzm. Dr. Ayşegül Büte Yüksel
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Endokrinoloji Bilim Dalı Olgu Sunumu 28 Şubat 2013 Uzm. Dr. Ayşegül Büte Yüksel

2
UZM.DR AYŞEGÜL BÜTE YÜKSEL
Sabah Vakası UZM.DR AYŞEGÜL BÜTE YÜKSEL

4
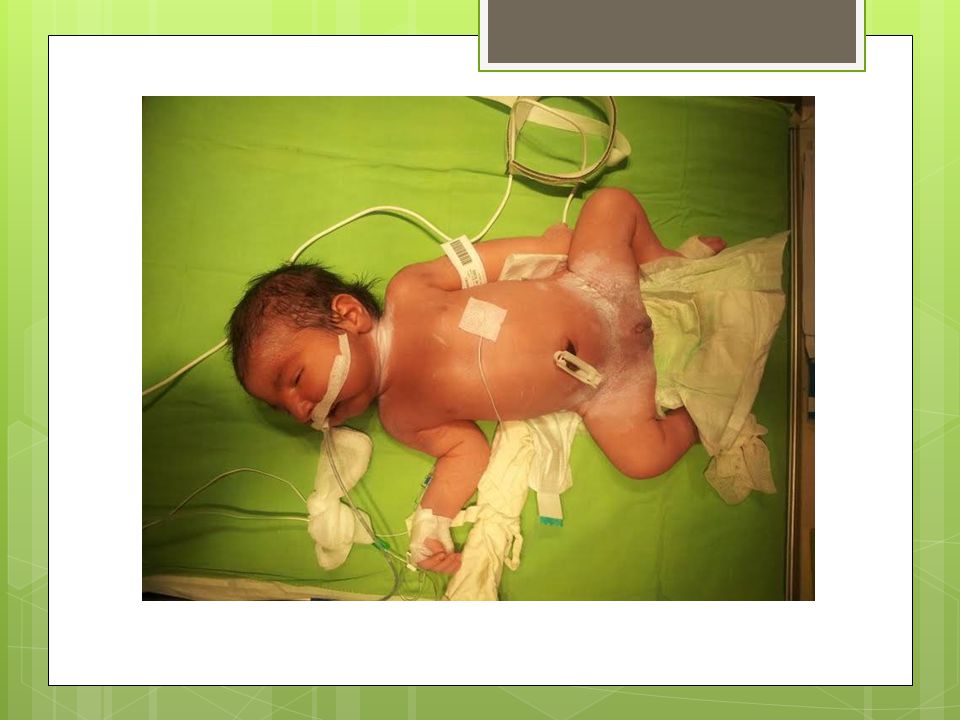
HİKAYESİ 3680 gr olarak NSD ile dış merkezde doğmuş,
Doğduğunda mekonyum ile boyalı ve sonrasında solunum sıkıntısı olan hasta mekonyum aspirasyon sendromu düşünülmesi nedeni ile YDYBU ne sevk edildi.

5
SOYGEÇMİŞ 23 yaşında sağlıklı anne; 35 yaşında sağlıklı babadan 2. yaşayan bebek, 1 tane intrauterin ex mevcut, Anne baba 1 .derece akraba(kuzen) Gebelik döneminde düzenli kontrolleri yapılmış, Gebeliğin son 20 gününde oligohidroamniyozu olmuş.
 Gebelik döneminde düzenli kontrolleri yapılmış, Gebeliğin son 20 gününde oligohidroamniyozu olmuş.")
9
FİZİK MUAYENE Genel durumu orta,bilinç açık,turgor normal, hipotonisi mevcut, Tartısı:3680 gr(10-25 per/-1,2 SDS) Boy:48 cm(<3.per/-2,26 SDS) Baş çevresi:33 cm (<3.per) Baş boyun bölgesinde atipik yüz görünümü , mikrosefalisi, mikroftalmisi, retrognatisi, düşük kulağı var, yükse damağı mevcut, Kardiavasküler muayenesinde özellik yok,
 Baş çevresi:33 cm (<3.per) Baş boyun bölgesinde atipik yüz görünümü , mikrosefalisi, mikroftalmisi, retrognatisi, düşük kulağı var, yükse damağı mevcut, Kardiavasküler muayenesinde özellik yok,")
13
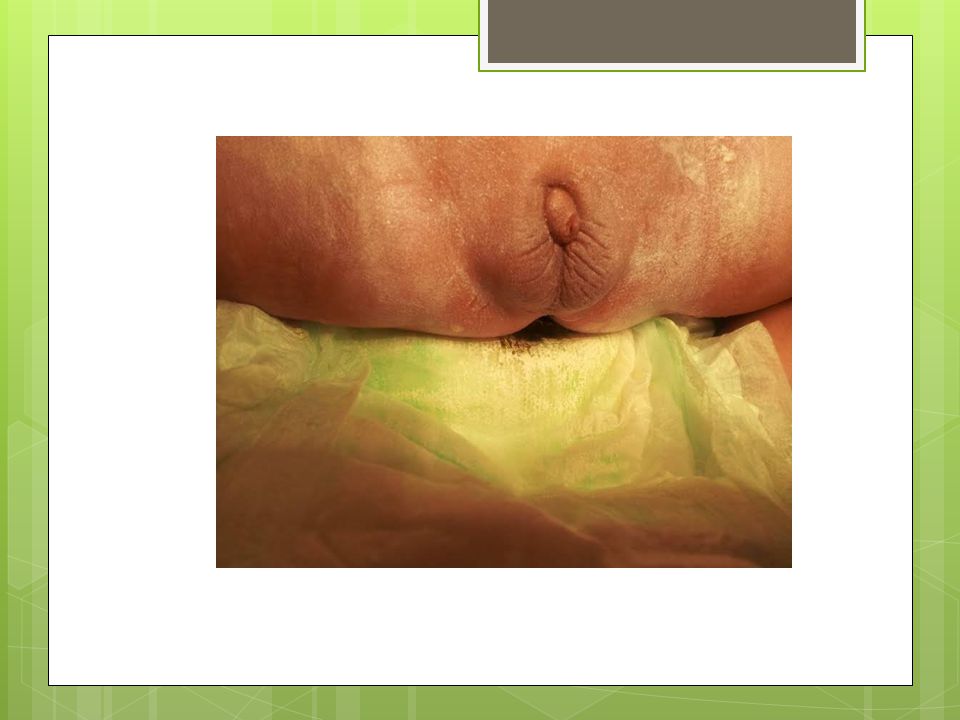
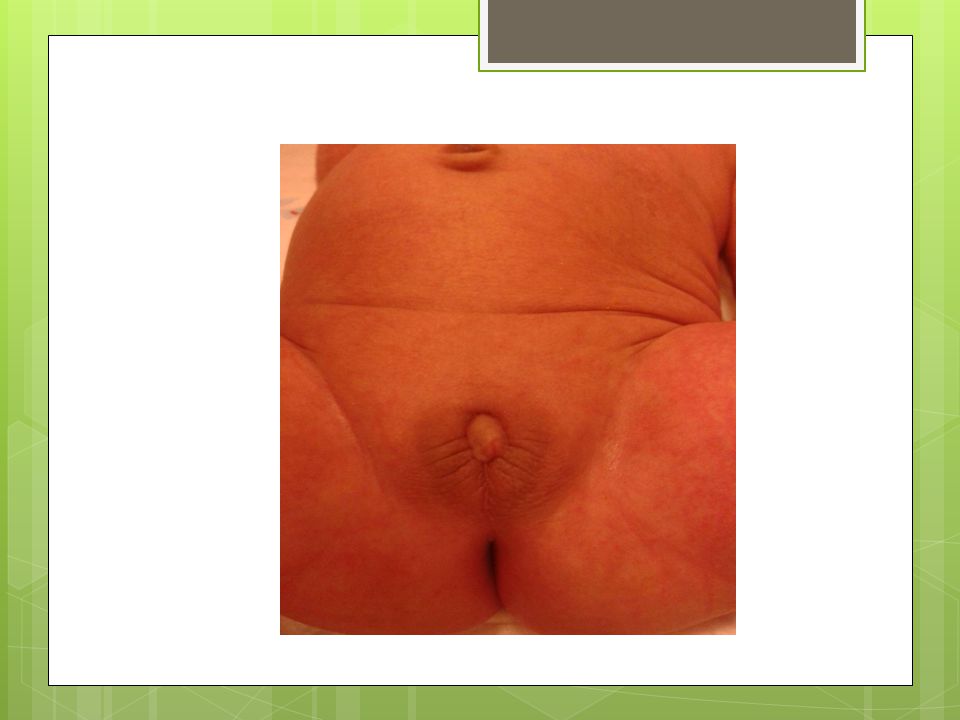
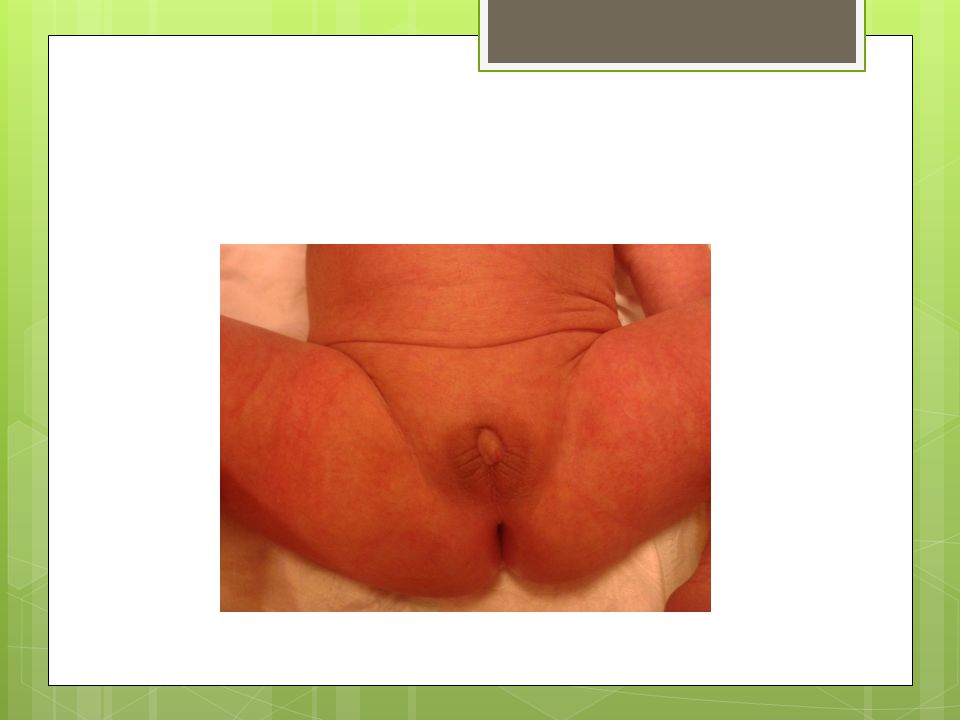
FİZİK MUAYENE Hastanın her iki ayak parmagında da polidaktilisi mevcut, solda kolda mora alınamadı. Genital bölge muayenesinde: Fallusu vardı ve 1 cm kadar ölçüldü., Scrotalize labium majusu var, Labium majuslar içinde bilateral palpe edilen gonadları vardı. Sacral bölgede kıllanma artışı mevcut.

14
RADYOLOJİ Abdominal USG:Bilateral grade 2 hidronefrozu var, parankim ekosu artmış.Uterus ve overler izlenmedi. Pelvik USG:İnguinal kanal distal ucunda olduğu düşünülen sağda 9*4mm, solda 10*5mm boyutunda solid görünümde testis ile uyumlu olabilecek dokular görüldü. Sacral USG:Normal EKO:PFO görüldü.

15
GÖZ MUAYENESİ Bilateral kataraktı vardı.

16
LABORATUAR 1.2.13 TSH 1,51 1,89 1,14 0,76 ST3 1,61 3,17 3,26 ST4 1,5 1,27 1,15 1,07 FSH 1,47 0,6 6,11 LH <0,2 0,39 0,21 DHEAS 73,3 T.TESTES 10,3 5,96 43,98 17.OH.P 1,64 1,46 1,42

17
LABORATUAR 7 dehidrokolesterol: 3,4 mmol/l (N:<7)
Kolesterol değerleri: TG:115 TK:122 HDL:24 LDL:75 VLDL:23

18
Patolojik bulgular Mikrosefali Hipotelorizm Mikrognati Katarakt
Polidaktili Yetersiz virilize male

19
ÖN TANINIZ NEDİR

20
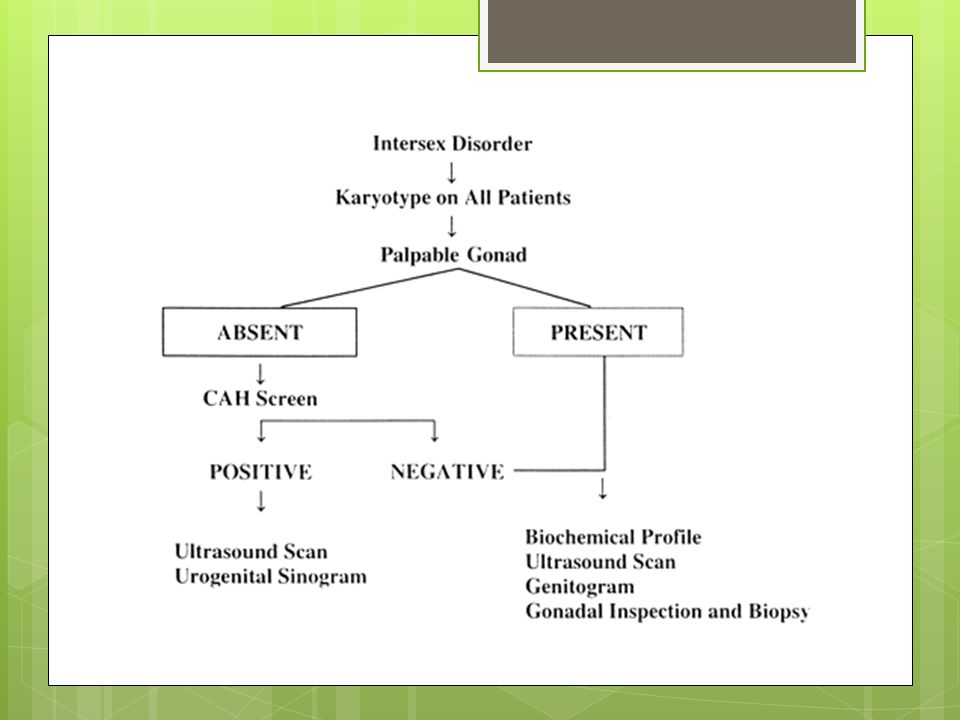
ÇOCUK ENDOKRİN DEĞERLENDİRMESİ-1
Hasta sendromik özellikleri ve kuşkulu genitalyası olması nedeniyle tarafımıza danışıldı. CGB düşünülen hastanın karyogramı, ayrıntılı pelvik ve batın USG si, babygramı, bazal FSH, LH, testesteron, DHEAS, 1-4 androstenidionu, 17 oh progesteron, kortizol, ACTH, AMH, lipitleri istendi. Polidaktilisi, kataraktı, CGB olması nedeni ile hastada ilk planda SLO sendromu düşünüldü ve 7 dehidrokolesterol tetkikide istendi.

21
Cinsel gelişim bozuklukları (CGB):Ambigius genitale/Intersex
Günümüzde cinsel gelişim bozukluğu-CGB (Disorder of sex development -DSD) olarak isimlendirilmektedir. Ambigius genitale, genital yapı gelişiminin çeşitli nedenlerle ve çoğunlukla androjenler üzerinden işleyen mekanizmalarda (kızlarda androjen fazlalığı, erkeklerde androjen azlığı gibi) anatomik eşitliklerin arasında kalması olarak tanımlanabilir
 olarak isimlendirilmektedir. Ambigius genitale, genital yapı gelişiminin çeşitli nedenlerle ve çoğunlukla androjenler üzerinden işleyen mekanizmalarda (kızlarda androjen fazlalığı, erkeklerde androjen azlığı gibi) anatomik eşitliklerin arasında kalması olarak tanımlanabilir.")
22
Şimdiki Disorder of sex development (DSD)/Cinsel gelişim bozukluğu (CGB) 46,XY CGB 46,XX CGB Ovatestiküler CGB 46,XX testiküler CGB 46, XY komplet gonadal disgenesis Önceki Intersex Male pseudohermafroditizm Yetersiz virilize XY erkek/ Yetersiz maskülinize XY erkek) Female pseudohermafroditizm Aşırı virilize XX kız Aşırı maskülinize XX kız Gerçek hermafroditizm XX erkek veya XX sex reversal XY sex reversal
 Female pseudohermafroditizm. Aşırı virilize XX kız. Aşırı maskülinize XX kız. Gerçek hermafroditizm. XX erkek veya XX sex reversal. XY sex reversal.")
23
SIK KULLANILAN TERİMLER
. Terimler Bifid skrotum: Skrotum iki parça gibi görünmesi Kliteromegali:Klitorisin büyümesi Hipospadyas: Üretra açıklığının penis ucundan aşağıda olması Mikrofallus: Normal yeni doğanda gerdirilmiş penis boyunun <2.5 cm olması Posterior Füzyon: Labiaların anüse doğru füzyon olması

24
CGB düşündüren klinik durumlar
Belirgin genital ambigius (cloacal exstrophy gibi) Belirgin Dişi Görünümle birlikte Büyük klitoris Posterior labial füzyon İngüinal/labial kitle (gonad) Belirgin Erkek görünümle birlikte Çift taraflı inmemiş testis Mikropenis İzole perianal hipospadyas Hafif hipospadyas+inmemiş testis Komplet androjen duyarsızlığı sendromu gibi ailesel CGB öyküsü Prenatal karyotip ile genital görünüm arasında uyumsuzluk
 Belirgin Dişi Görünümle birlikte. Büyük klitoris. Posterior labial füzyon. İngüinal/labial kitle (gonad) Belirgin Erkek görünümle birlikte. Çift taraflı inmemiş testis. Mikropenis. İzole perianal hipospadyas. Hafif hipospadyas+inmemiş testis. Komplet androjen duyarsızlığı sendromu gibi ailesel CGB öyküsü. Prenatal karyotip ile genital görünüm arasında uyumsuzluk.")
25
Cinsel farklılaşma adımları:

26
Etyolojik sınıflama Artmış virilizasyon Maskülinizasyon yetersizliği
46 XX androjen fazlalığı (KAH) Aromataz eksikliği İatrojenik Maskülinizasyon yetersizliği 46 XY gonadal disgenesiz(45,X/46,XY dahil) Andojen biyosentez bozuklukları ( KAH ile birlikte veya değil) Androjen duyarsızlığı Klasifiye edilemeyen hipospadyas Gerçek hermofraditizm Non-endokrin malformasyonlar (Epispadyas sendromları)
 Aromataz eksikliği. İatrojenik. Maskülinizasyon yetersizliği. 46 XY gonadal disgenesiz(45,X/46,XY dahil) Andojen biyosentez bozuklukları ( KAH ile birlikte veya değil) Androjen duyarsızlığı. Klasifiye edilemeyen hipospadyas. Gerçek hermofraditizm. Non-endokrin malformasyonlar (Epispadyas sendromları)")
27
CGB sınıflaması: 47,XXY( Klinefelter sendromu ve varyantları
46, XY CGB Gonadal (testiküler) gelişim bozuklukları 1.Komplet gonadal disgenesis (Swyer send) 2.Parsiyel gonadal disgenesis 3.Gonadal regresyon 4.Ovotestiküler CGB Androjen sentez veya etkisindeki bozukluklar 1.Androjen biyosentez defektleri (17-OHSD,5-alfa redüktaz vb) 2.Androjen etkisinde defekt ( CAIS, PAIS) 3. LH reseptör defekti (leidig hücre aplazi/hipoplazisi 4.AMH, AMH reseptör defekti (Persistan mullerian duct send) (C) Diğer (Şiddetli hipospadyas, klaokal ekstrofi) 46, XX CGB Gonadal (Ovaryan) gelişim bozuklukları 1.Ovatestiküler CGB 2.Testiküler CGB ( SRY +) 3.Gonadal disgenesis (B) Androjen fazlalığı 1. Fetal (21 OH az, 11-OH az eksikliği) 2. Fetoplasental (Aromataz eksikliği, POR) 3.Maternal (Lutemo, eksojen vs) (C) Diğer(klaokal ekstrofi, vaginal atrezi, MURCS) Sex kromozom CGB 45,X (Turner sendromu ve varyantları) 47,XXY( Klinefelter sendromu ve varyantları 45, X/46,XY( miks gonadal disgenesis, ovotestiküler CGB) 46, XX/46, XY (Chimeric, ovotestiküler CGB
 gelişim bozuklukları. 1.Komplet gonadal disgenesis (Swyer send) 2.Parsiyel gonadal disgenesis. 3.Gonadal regresyon. 4.Ovotestiküler CGB. Androjen sentez veya etkisindeki bozukluklar. 1.Androjen biyosentez defektleri (17-OHSD,5-alfa redüktaz vb) 2.Androjen etkisinde defekt ( CAIS, PAIS) 3. LH reseptör defekti (leidig hücre aplazi/hipoplazisi. 4.AMH, AMH reseptör defekti (Persistan mullerian duct send) (C) Diğer (Şiddetli hipospadyas, klaokal ekstrofi) 46, XX CGB. Gonadal (Ovaryan) gelişim bozuklukları. 1.Ovatestiküler CGB. 2.Testiküler CGB ( SRY +) 3.Gonadal disgenesis. (B) Androjen fazlalığı. 1. Fetal (21 OH az, 11-OH az eksikliği) 2. Fetoplasental (Aromataz eksikliği, POR) 3.Maternal (Lutemo, eksojen vs) (C) Diğer(klaokal ekstrofi, vaginal atrezi, MURCS) Sex kromozom CGB. 45,X (Turner sendromu ve varyantları) 47,XXY( Klinefelter sendromu ve varyantları. 45, X/46,XY( miks gonadal disgenesis, ovotestiküler CGB) 46, XX/46, XY (Chimeric, ovotestiküler CGB.")
28
Virilizasyon(maskülinizasyon) yetersizliği
46 XY gonadal disgenesiz(45,X/46,XY dahil) Andojen biyosentez bozuklukları ( KAH ile birlikte veya değil) Androjen duyarsızlığı Klasifiye edilemeyen hipospadyas
 Andojen biyosentez bozuklukları ( KAH ile birlikte veya değil) Androjen duyarsızlığı. Klasifiye edilemeyen hipospadyas.")
29
Testesteron sentez bozuklukları
5α reductase 17 hydroxy steroid dehydrogenase 17 – 20 lyase 3β hydroxy steroid 17 hydroxylase LH (hCG) × Leydig Cell Hypoplasia StAR protein deficiency Pregnenolone 17 hydroxy- DHEAS Androstenedione Testosterone Dihydrotestosterone Androgen receptor Cholesterol × × × Androgen Insensitivity
 × Leydig Cell Hypoplasia. StAR. protein. deficiency. Pregnenolone. 17 hydroxy- DHEAS. Androstenedione. Testosterone. Dihydrotestosterone. Androgen receptor. Cholesterol. × × × Androgen Insensitivity.")
30
5α-Redüktaz eksikliği Periferik dokularda DHT sentezinde bozukluk olur
Sıklıkla dişi fenotip+parsiyel virilizasyon ama tam dişi görünümü de olabilir Uterus ve mullerien yapılar yoktur Testis sağlam ve yeterli T salgılar Pubertede virilizasyon olur Erkek olarak fertildirler ve kız cinsel kimliği verilse bile puberte erkek kimliğine geri dönme ihitimalleri yüksektir

31
46 XY genotipinde, normal gonad gelişimini tamamlamış erkek cinsiyette intrauterin dönemde genital virilizasyonu DHT tarafından sağlanmaktadır. Testesteron 5alfa redüktaz enzimi ile DHT a irreversibl olarak dönüştürülür. SRD5A1 ve SRD5A2 genleri 5alfa redüktazı sentezleyen genlerdir. 46 XY infantta SRD5A2 geninde oluşucak mutasyonlar sonucunda hastada izole hipospadiasdan ciddi maskulinizasyon sorunlarına kadar ciddi CGB oluşabilir.

32
5alfa redüktaz tanısı infant yada puberte döneminde bazal yada BHCG uyarısı sonrasında T/DHT oranının artması ile konulabilir. Yd da T/DHT>8,5 olması; Cocukluk ve pubertal dönemdede sınır 10 olarak alınmaktadır.Ancak >10 olması tanıyı desteklesede; <10 olması tanıyı dışlamamaktadır.

33
Testesteron sentez kusuru:
Kolesterolden testesteron yapımı sırasındaki enzimatik kusurlar 46XY CGB neden olabilir. OR geçişlidir. Kuşkulu genital yapı ile beraber Smith-Lemli-Opitz ve Ankley-Bixler gibi sendromlarda iskelet, kalp, böbrek gibi sistemlerinde tutulması ile baraber komplek bir fenotip ortaya çıkabilir. Bu hastalarda genetik danışma gereklidir.

34
Smith-Lemli-Opitz Sendromu
7-dehidrokolesterolden (7DHC) kolesterol redüktaz enzimi ile kolesterol sentezlenmektedir.Bu hastalarda bu enzim eksikliği nedeni ile sentez yapılamaz. 7DHC miktarı kanda artar ve hastanın klinik bulgularının dereceside bu durum ile doğru orantılıdır. Bu hastalarda tipik yüz görünümü,yarık damak, kalp, böbrek, gis ve ac anomalileri, boy kısalığı, zeka geriliği, 2-3.parmaklar arasında sindaktili,hipotoni ve kuşkulu genitalya görülmektedir. Tedavisi kolesterol verilmesidir.
 kolesterol redüktaz enzimi ile kolesterol sentezlenmektedir.Bu hastalarda bu enzim eksikliği nedeni ile sentez yapılamaz. 7DHC miktarı kanda artar ve hastanın klinik bulgularının dereceside bu durum ile doğru orantılıdır. Bu hastalarda tipik yüz görünümü,yarık damak, kalp, böbrek, gis ve ac anomalileri, boy kısalığı, zeka geriliği, 2-3.parmaklar arasında sindaktili,hipotoni ve kuşkulu genitalya görülmektedir. Tedavisi kolesterol verilmesidir.")
35
Androjen duyarsızlığı sendromu( AIS)
46 XY CGB ‘larının en sık nedenidir. Androjen rsp gen mutasyonlarına bağlı gelişen bir sendromdur. X ‘e bağlı seyreder. Hem endojen hem eksojen androjen direnci vardır. Direncin derecesine göre komplet, parsiyel, hafif formu olabilir. Fenotip normal bir kızdan fertil erkeğe kadar değişebilir.

36
Androjen duyarsızlığı sendromu( AIS)
Genotip: 46 XY Komplet/inkomplet olabilir Komplet (testiküler feminizasyon) Dış genital tamamen kızdır. İç genital yapılar (kız veya erkek) yoktur ingüinal veya labial kitle(testis) Amenore İnkomplet Değişik derecelerde ambigius, ingüinal veya labial kitle(testis), Diğer bakımlardan normal erkeklerde infertilite/ jinekomasti LH, FSH ve Testesteron yüksek
 Dış genital tamamen kızdır. İç genital yapılar (kız veya erkek) yoktur. ingüinal veya labial kitle(testis) Amenore. İnkomplet. Değişik derecelerde ambigius, ingüinal veya labial kitle(testis), Diğer bakımlardan normal erkeklerde infertilite/ jinekomasti. LH, FSH ve Testesteron yüksek.")
38
ÇOCUK ENDOKRİN DEĞERLENDİRMESİ-2
46 XY CGB düşünülen hastaya 26 günlükken başlanarak 3 gün 1500 mcg B-HCG ile uyarı testi yapıldıktan sonra hastadan testesteron ve dihidrotestesteron alındı, henüz sonuçları çıkmadı. Hastadan karyogram gönderilmişti, sonucu bekleniyor.

39
Hasta ilk 19 gününde YDYBÜ de takip edildi
Hasta ilk 19 gününde YDYBÜ de takip edildi. Solunum sıkıntısı gerileyen hasta serviste anne yanında ızlenmeye devam edildi ve 29 günlükken taburcu edildi.Yenidoğan polikliniğinde izlemine devam ediliyor. Göz hastalıkları ile katarakt ameliyatı planlandı. Çocuk nefroloji polikliniğinde takibine alındı. Çocuk nöroloji ile konsulte edildi, hastadan Tandem MS, idrarda organik asit, kongenital glikolizasyon defektleri açısından kan ve idrar tetkikleri alındı. KLİNİK SEYİR

40
TEŞEKKÜRLER!!!!












>")








>")


