Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
AMİNO ASİT METABOLİZMASI BOZUKLUKLARI

2
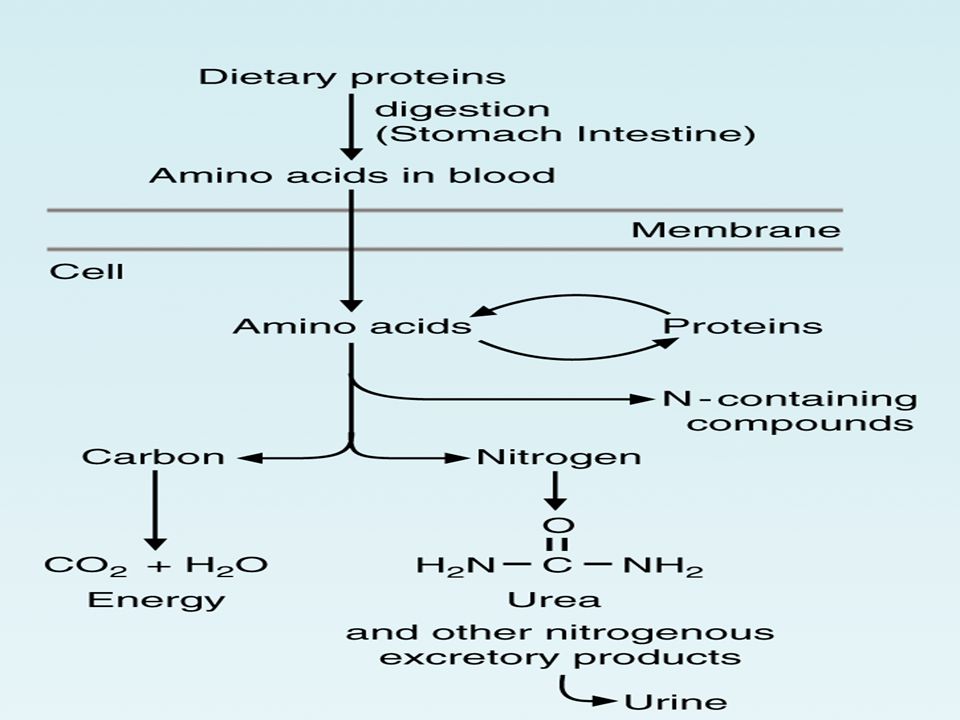
Sindirim kanalında besinsel proteinlerin enzimlerin etkisiyle parçalanması sonucu amino asitler oluşur ve bunlar emilerek vücuda alınırlar. Besinlerden kaynaklanan amino asitlere ek olarak önemli miktarda amino asit doku metabolizması sırasında sentezlenmektedir.

3
Hücreler içindeki proteinler devamlı olarak yıkılmakta ve yeniden sentezlenmektedir.
VÜCUT PROTEİNİ Protein dönüşümü sırasında serbest hale geçen amino asitlerin %75-80’i yeni protein sentezi için tekrar kullanılır. Amino asitler

5
Çeşitli kalıtsal hastalıklar, A
Çeşitli kalıtsal hastalıklar, A.A metabolizmasındaki bozukluklardan oluşur. Bu bozukluklar A.A’rin normal metabolizmalarında görevli anahtar bir enzimin eksikliği veya bloku söz konusudur. Özel önem taşıyan A.A şunlardır; Fenilalanin, tirozin, sistin; homosistein ve diğer dallı zincirli A.A’dir.

7
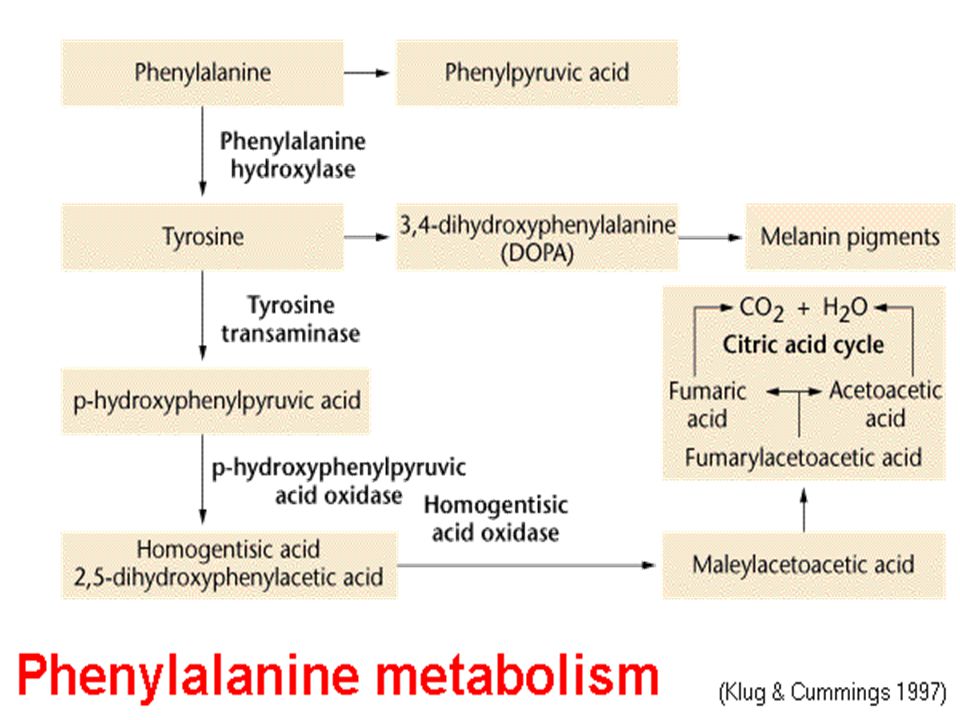
Fenilalanin Metabolizması Bozuklukları
Fenilalanin kan düzeyleri 2 mg/dL’den daha yüksektir. Normal kişilerde fenilalanin hidroksilaz fenilalanini tirozine hidroksiller. Hiperfenilalaninemiler bu enzimin defektine bağlı olarak meydana gelir

8
Klasik Fenilketonüri (PKU):
Amino asit metabolizmasının en sık karşılaşılan hastalığı, dünya da yaklaşık yenidoğandan birinde görülmektedir. Otozomal resesif taşınmakta İsmini idrarda yüksek miktarda bulunan fenilketondan almaktadır

9
Klasik Fenilketonüri

10
Büyük çapta fenilalanin hidroksilaz eksikliğine bağlı olmakla birlikte;
Daha nadir olarak tetrahidrobiopterin kofaktörünün veya kofaktörün indirgenmiş aktif durumda kalmasını sağlayan dihidrobiopterin redüktaz eksikliğine bağlı olarak ortaya çıkmaktadır. Fenilalanin hidroksilaz kusuru tam (klasik PKU) veya kısmi (tip ll ve lll)olabilmektedir.
 veya kısmi (tip ll ve lll)olabilmektedir.")
11
Tüm olguların yaklaşık yarısını oluşturan klasik PKU de fenilalanin hidroksilaz hiç bulunmamakta ve normal şekilde katabolize olamayan fenilalanin tüm vücut sıvılarında birikmektedir. Normalde <%2 mg olan erişkin pha düzeyi, klasik tipte >%20 mg olabilmektedir Fenilalanin, fenilpiruvat üzerinden katabolize olduğu için idrar ile büyük miktarda fenilpiruvat ile bu ketonun dönüştüğü fenilasetat, fenilasetilglutamin artmaktadır

12
Doğumda normal görünen bebeklerde kısa zaman içinde gelişme geriliği, beslenme güçlüğü ve kusma belirtileri ortaya çıkar. Artmış fenilpiruvat nedeni ile bazı bebeklerin idrar ve terlerinde küf kokusu olmaktadır. Fenilalanin ve metabolitlerinin birikimi beyin gelişimini bozarak zeka geriliğine yol açmaktadır. Beyinde gelişen kusurlu miyelinizasyon yüzünden sık epileptik nöbetler görülmektedir

13
Fenilketonların beyinde serotonin enzimatik sentezini engellemelerine bağlı olarak düşük serotonin düzeyinin bu hastalardaki zeka kusurlarının nedeni olabilir Ayrıca fenilglutaminin oluşumunda fazla glutamin kullanılmasına bağlı olarak , kronik glutamin azlığının beyin hasarında doğrudan etkili olabileceği ileri sürülmektedir FKU’li çocukların % 60’ında açık saç rengi, açık göz rengi, açık cilt rengi ile karakterize görünümü vardır

14
Fenilketonüride erken tanı önemlidir.
Çünkü besinsel tirozin, endojen tirozin sentez eksikliğini ortadan kaldırmada yeterli görülmemektedir. Fenilketonüride erken tanı önemlidir. Dolayısıyla neonatal dönemde kan fenilalanin düzeyinin ölçülmesi önemlidir En yaygın kullanılan ve yarı kantitatif mikrobiyolojik yöntem olan Gutrie testi dir. * Test pozitif ise daha ileri tetkikler olan kan ve idrardır.

15
PKU tedavisi düşük proteinli besinler ile yapılmaktadır
PKU tedavisi düşük proteinli besinler ile yapılmaktadır. Geri dönüşümü olmayan beyin hasarını önlemek için düşük proteinli besin tedavisinin doğumdan hemen sonra başlaması ve bu tedavinin 8-10 Yaşa kadar ciddi bir şekilde devam etmesi gerekmektedir (yaşam boyu fenilalaninden kısıtlı diyet). Biopterin ve biopterin redüktaz eksikliğinin tedavisi, fenilalanin ve tirozin kısıtlılığı ile çözülmez (sentezlenemeyen öncül bileşikler verilmeli).
. Biopterin ve biopterin redüktaz eksikliğinin tedavisi, fenilalanin ve tirozin kısıtlılığı ile çözülmez (sentezlenemeyen öncül bileşikler verilmeli).")
16
Geçici neonatal hiperalaninemi
Kalıtsal bir eksiklik olmayan bu bozukluk, karaciğerin olgunlaşmasının gecikmesine bağlı olarak fenilalanin enzim sisteminin yetersizliğidir. Bebek büyüdükçe fenilalanin düzeyi normal sınırlar içinde kalır. Tirozinemiler Tip ll tirozinemi: tirozin transaminaz eksikliği, hipertirozinemi ve tiroziüri ile ortaya çıkar.

17
İdrarda n-asetiltirozin, hidroksifenil piruvat, hidroksifenil laktat ve hidroksi fenilasetat gibi tirozin metabolitlerinin atılımı artmaktadır. Göz ve deri lezyonları, tirozinin, intaselüler kristaleşmesine bağlı olan inflamasyon ve birçok olguda görülen zeka geriliği ile karakterize bir hastalıktır.

18
Tip l tirozinemi(hepatorenal tirozinemi).
Fumarilasetoasetat hidroksilaz eksikliğine bağlıdır. Karaciğer yetmezliği, renal tübüler fonksiyon bozukluğu, anemi ve D-vitaminine dirençli raşitizim ile birlikte, idrar ile tirozin, metiyonin gibi amino asitler ve metabolitleri atılmaktadır. Besinsel kısıtlama, ilerleyici karaciğer hasarını iyileştirememektedir.

19
Neonatal tirozinemi Bebeklikte tirozinemilerin en sık görülen tipidir, özellikle prematür bebelerde Kc’in olgunlaşmamasından dolayı serum tirozin yüksektir, geçici bir durumdur, ve hidroksi-fenilpiruvat oksidaz eksikliği sonucu ortaya çıktığı düşünülmektedir. Karaciğerin olgunlaşması ile (4-8 hafta) normal dönüş olur.
 normal dönüş olur.")
20
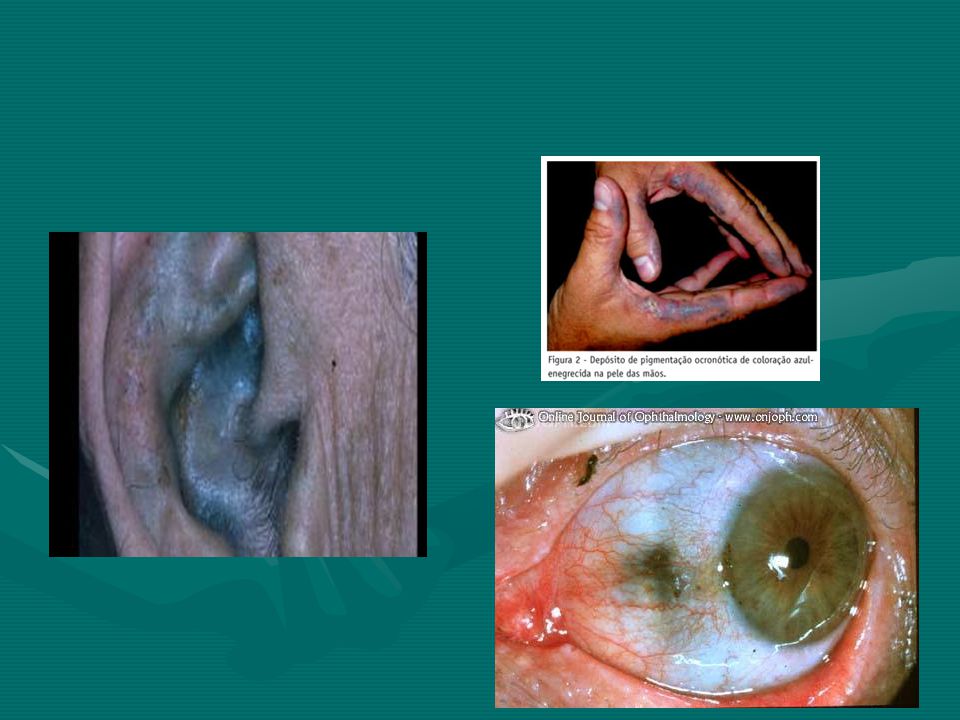
Alkaptonüri: (ilk tanımlanan kalıtsal metabolik hastalık, nadir gözüken), homogentizik asit oksidaz (HGA oksidaz) eksikliğine bağlı olarak homogentizik asit birikmekte ve idrarla büyük miktarda atılmaktadır. Renksiz bir hidrokinon olan bu bileşik, beklediği zaman oksidasyon ve polimerizasyon ile koyu renkli melanine benzeyen bir pigment oluşturmaktadır. Alkaptonürinin en çarpıcı klinik belirtisi idrar renginin açık havada bekletilmekle esmerleşmesidir.

21
Hastalığın geç evrelerinde oksidlenmiş homogentizik asit kıkırdak, kemikler ve diğer organlarda birikmesi sonucu görülen yaygın pigmantasyona okronozis adı verilmektedir. Özellikle erkeklerde pigment depolanması artirit gelişimine neden olmaktadır. Tirozin ve fenilalanin kısıtlanması yararlı olmaktadır (ancak tatmin edici bir tedavi yoktur). Ayrıca homogentisik asit oksidazın maksimum aktivitesi için C vitamini de verilmektedir.
. Ayrıca homogentisik asit oksidazın maksimum aktivitesi için C vitamini de verilmektedir.")
23
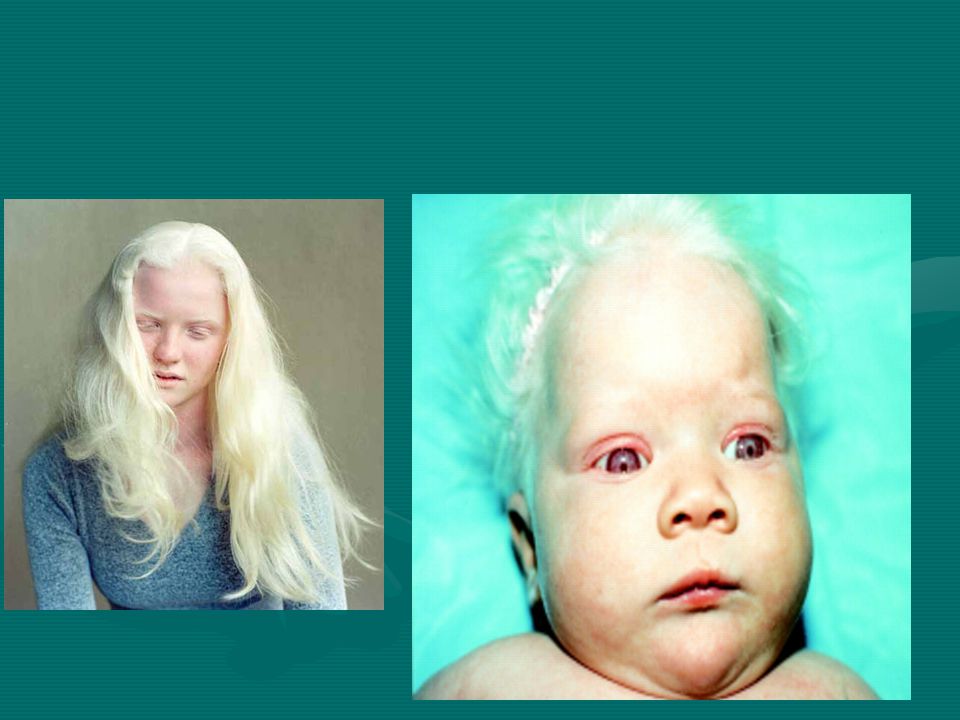
Albinizim: Melanin sentezindeki kalıtsal hatalara bağlı olarak ortaya çıkan bir sendrom.
Tirozinaz enziminin (melanositlerde tirozinin melanine dönüşümünü sağlayan bakır içeren bir enzim) yokluğu veya yetersizliğine bağlı olarak deri, saç ve gözlerde pigmentasyon azalmaktadır. Deride pigment azlığı , albinoların güneşe duyarlılığını artırmakta ve sıklıkla deri kanserlerine yol açmaktadır. İris pigmantasyonunun azalması, fotofobiye neden olmaktadır.
 yokluğu veya yetersizliğine bağlı olarak deri, saç ve gözlerde pigmentasyon azalmaktadır. Deride pigment azlığı , albinoların güneşe duyarlılığını artırmakta ve sıklıkla deri kanserlerine yol açmaktadır. İris pigmantasyonunun azalması, fotofobiye neden olmaktadır.")
25
Sistinüri En sık rastlanan doğuştan A.A transportu bozukluğudur.
İdrarla aşırı miktarda sistin, lizin, arjinin, ve ornitin atılır. Bu A.A ler için spesifik taşıyıcılar vardır, bunlardan birincisi sistin, ikincisi lizin arjinin ornitin, üçüncüsü bu dört A.A’ din tümü içindir. Sistinüri üçüncü taşıyıcı sistemin defektine bağlı oluşur.

26
Sistin en az çözünen A.A’dir, idrarla aşırı atılımı renal pelvis, üreterler ve mesanede sistin taşlarının oluşumuna yol açar. Tedavide hastaya bol su içirilerek sistinin idrardaki konsantrasyonunun düşmesi amaçlanır.

27
Homosistinüri/Homosisteinemi
Homosistein, metiyonin sistein metabolik yolunda önemli dallanma noktasındadır. Karasız bir molekül olduğundan homosistein normalde plazmada birikmez. Homosisteinemi kardiovasküler hastalıkla ilişkilendirilmektedir ve yüksek kan düzeylerinin önemli bir risk faktörü olduğu düşünülmektedir

28
Homosistinüri Görülme insidansı 1: Vücut dokularında homosistein konst. artışı ile karakterizedir

29
Maple Syrup Hastalığı (MSUD)
İsmini hastalıklı kişilerin idrarının karakteristik kokusundan almaktadır. Akçaağaç şurubu veya yanmış şeker kokusu, idrarda alifatik keto asitlerin yüksek konst. Olmalarından kaynaklanmaktadır.. Kan ve idrar örneklerinde, A.A analizi yüksek düzeyde lösin, izolösin ve valin gösterir. Klasik tipte, hastalıklı bebekler doğumda normal görünürler, daha sonra sık kusma ve gelişme geriliği görülür. Ciddi nörolojik fonksiyon bozukluğu, nöbet, koma, solunum yetmezliği ve birçok hastada ölüme yol açar. Hayatta kalan hastalar genellikle zihinsel gerilik gösterir.

30
Tietz, Klinik Kimyada Temel İlkeler
Lippincott Marks’ Temel Tıbbi Biyokimyası, Klinik Yaklaşım

Benzer bir sunumlar