Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
KALSİYOTROPİK İLAÇLAR Parathormon, D Vitamini ve Kalsitonin
Prof. Dr. Hakan KARADAĞ

2
Kalsiyum % 99'u kemiklerde hidroksiapatit şeklinde bağlı durumdadır. Ca10(PO4)6(OH)2 Plazma düzeyi: 10 mg/dL (5 mEq/L) (2,5 mmol/L) İyonize kalsiyum (% 50) Proteine bağlı kalsiyum (% 40) % 90 albumin Kalanı globulinlere bağlıdır. Proteine bağlanma pH'a bağımlıdır. pH (asidoz): bağlanma azalır, iyonize Ca2+ pH (alkaloz): bağlanma artar, iyonize Ca2+ Suda çözünen kompleks şeklinde (% 10)
 Proteine bağlı kalsiyum (% 40) % 90 albumin Kalanı globulinlere bağlıdır. Proteine bağlanma pH a bağımlıdır. pH (asidoz): bağlanma azalır, iyonize Ca2+ pH (alkaloz): bağlanma artar, iyonize Ca2+ Suda çözünen kompleks şeklinde (% 10)")
3
Sinir, kas ve diğer eksitabl hücrelerin sitoplazma membranının Na+ ve K+'a permeabilitesinde serbest Ca2+ düzeyi önemli rol oynar Ca2+ membrandaki Na+ kanalları daha kolay ve daha fazla açılır stimülasyon eşiği , eksitabilite artar spontan depolarizasyonlar gelişebilir (örn. hipokalsemik tetani)
")
4
Kalsiyum dengesinde 3 organın önemli fizyolojik rolleri vardır
Barsaklar Aktif transport kalsiyum bağlayan protein (CaBP) kalbindin Kısmen pasif difüzyon Kemikler Osteoblastlar Osteoklastlar Böbrekler glomerüler filtrasyon
 kalbindin. Kısmen pasif difüzyon. Kemikler. Osteoblastlar. Osteoklastlar. Böbrekler. glomerüler filtrasyon.")
6
Estrojenler (bazı sitokinlerin üretimini azaltarak) (?) IL-1 IL-6 TNFa
Osteoklastlar Stimüle edenler PTH 1,25(OH)2D3 IL-6, IL-11 İnhibe edenler Kalsitonin TGF-b IFNa PGE2 Estrojenler (bazı sitokinlerin üretimini azaltarak) (?) IL-1 IL-6 TNFa Dolaylı yoldan stimülasyon Osteoblastlar Stimüle edenler PTH 1,25(OH)2D3 IL-1 hGH, IGF1 PGE2 TNF Estrojenler (?) İnhibe edenler Kortikosteroidler Dvit-R PTH-R Kalsitonin-R Osteoblast + - Osteoklast Monosit
2D3. IL-6, IL-11. İnhibe edenler. Kalsitonin. TGF-b. IFNa. PGE2. Estrojenler (bazı sitokinlerin. üretimini azaltarak) ( ) IL-1. IL-6. TNFa. Dolaylı yoldan stimülasyon. Osteoblastlar. Stimüle edenler. PTH. 1,25(OH)2D3. IL-1. hGH, IGF1. PGE2. TNF. Estrojenler ( ) İnhibe edenler. Kortikosteroidler. Dvit-R. PTH-R. Kalsitonin-R. Osteoblast. + - Osteoklast. Monosit.")
7
Raşitizm Osteomalasi Kemik matriksinde bulunan kalsiyumun (mineral) azalması Osteoporoz Kemik matriksi ve mineralinin azalması

8
Normal Kemik Osteoporotik Kemik PATHOGENESIS OF OSTEOPOROSIS
INTRODUCTION – Osteoporosis is a skeletal disorder characterized by two elements that distinguish it from other causes of osteopenia such as hyperparathyroidism and osteomalacia: low bone mass; and microarchitectural disruption (show table 1) [1]. • Low bone mass is a characteristic finding in osteoporosis. Figure 1 shows normal bone in panel A, and progressively more osteoporotic bone in panels B, C and D (show figure 1). The bone that is present is normally mineralized, which distinguishes osteoporosis from osteomalacia. • There is disruption of the normal architecture, as illustrated in Figure 1, panel D (show figure 1). There are fewer bony spicules in osteoporotic bone and they are thinner than normal; in addition, there are horizontal "struts" that do not join up to any other structure, and thereby provide no structural support. This microarchitectural disruption undermines the structural integrity of the bone, and leads to the major clinical features of osteoporosis: skeletal fragility and an increase in fracture risk [1]. The mechanisms of the microarchitectural disruption are not clear. Increased remodeling itself may cause structural weakening, which may account for the independent association of high bone turnover with fracture risk (show figure 2) [2]. Other possible factors include: • Microfractures and fatigue damage • The development of perforations and discontinuities in trabecular bone, as well as a relatively excessive loss of horizontal trabeculae (show figure 1) • "Macroarchitecture" may play a role; as an example, increased length of the femoral neck appears to increase the risk of hip fracture • Posture, muscle strength and the frequency and type of falls affect fracture frequency and site. Decreased bone mass can occur because peak bone mass is low, bone resorption is excessive, or bone formation during remodeling is decreased. All three processes are likely to contribute, in varying degrees, to osteoporosis in individual patients. Their relative contribution to fracture risk is not known, but it seems likely that increased bone resorption has the greatest impact [3,4]. Age- and menopause-related bone loss are clearly important pathogenetic factors, but their expression must vary because there are wide variations in the amount of bone and the amount of "porosity" of bone in older persons of the same age (show figure 3) [5]. This topic will focus on the determinants of bone mass and remodeling that are likely to be important in the pathogenesis of decreased bone density. The epidemiology and causes of osteoporosis and the factors that normally regulate bone formation and resorption are discussed separately. (See "Epidemiology and causes of osteoporosis" and see "Normal skeletal development and regulation of bone formation and resorption"). DETERMINANTS OF PEAK BONE MASS – Based upon twin studies, genetic determinants account for 40 to 80 percent of the differences in peak bone mass. Skeletal structure and bone turnover are probably also genetically determined, but environmental factors affect bone growth during childhood and adolescence. Thus, increasing calcium intake and physical activity have a small positive affect on peak bone mineral density, moreover the increment in peak bone mass that occurs due to these lifestyle factors may have a substantial affect on the later incidence of fragility fractures [6]. As examples, calcium-enriched foods or supplements (approximately 1600 mg/day) promoted accrual of bone in children [7,8]. The effect lasted three to five years after the end of the supplementation in one, but not another study [9,10] (See "Calcium requirements in adolescents"). There may be important interactions between genetic and environmental factors. As an example, the differences in bone mass attributed to different alleles of the vitamin D receptor may be dependent upon differences in calcium intake [11]. Many genes have been examined for their possible role in the pathogenesis of osteoporosis [12,13]. • Different alleles of the gene for the vitamin D receptor are associated with small differences in bone mass [11]. However, the differences are not consistent among studies, and there is little evidence for a general association between vitamin D receptor alleles and osteoporosis [12,13]. • Increased frequency of osteoporosis has been reported in patients with a particular polymorphism of an Sp-1 cleavage site in the first intron of the collagen gene [12,13]. • Other genes, including those for the estrogen receptor, transforming growth factor-beta, and apolipoprotein E, have also been implicated in osteoporosis. • A report based upon an extensive linkage analysis suggested that polymorphisms in the bone morphogenetic protein-2 (BMP-2) gene were associated with an increased risk of fracture and low BMD in both pre- and postmenopausal women [14]. • An activating mutation of the gene for a low-density-lipoprotein (LDL) receptor-related protein 5 (LRP-5) was associated with high bone mass as an autosomal dominant in several families [15,16]. Transgenic mice carrying the activating mutant have increased bone mass and strength. Deletion of the LRP-5 gene causes an unusual autosomal recessive disorder, osteoporosis-pseudoglioma syndrome, in which bone mass is markedly reduced [17]. Whether alterations in the expression or activity of this gene and related signal transduction molecules are important in the pathogenesis of osteoporosis is not known. Not enough is known about the genetics of osteoporosis to affect treatment. The selection of patients for diagnostic work-up and preventive therapy could be improved if there were good genetic markers for risk. At least, some of the variability in the clinical course of osteoporosis might be explained. MECHANISMS OF BONE LOSS IN OSTEOPOROSIS – Bone loss can occur because bone resorption is increased or bone formation is decreased. There is considerable evidence that osteoporosis is associated with increased bone resorption, consistent with the morphologic pattern of trabecular bone loss and increased cortical porosity. However, bone biopsies in patients with osteoporosis do not clearly show an increase in actively resorbing surfaces [18]. Therefore, as compared with bone resorption, there must be a relative decrease in bone formation to account for bone loss. An increase in activation frequency, ie, in the number of bone-resorbing sites, leads to a transient decrease in bone mass. In normal subjects, there is a compensatory increase in bone formation, leading to restoration of bone mass. However, if the increase in resorption results in perforations of trabecular plates or discontinuities of trabecular struts, the template for new bone formation is lost and the decrease in bone mass is irreversible. The term "high turnover" osteoporosis has been used to describe patients in whom excessive bone resorption predominates, while "low turnover" has been applied to patients in whom defective formation predominates. However, the relative contributions of increased resorption and decreased formation probably represent a continuum, and this relation may change during the course of the disease. Biopsies of patients with advanced osteoporosis usually show decreased osteoblastic activity, but this may be an end stage of a process that began with excessive resorption. ROLE OF SYSTEMIC HORMONES IN OSTEOPOROSIS – Age- and menopause-related changes in the production of many hormones have been identified, but there are no differences in the serum concentrations of any hormone in patients with osteoporosis and suitably matched control subjects. Nevertheless, the changes in calcium-regulating hormones, sex hormones, and growth-regulating hormones that occur with age probably contribute to osteoporosis or at least result in increased susceptibility to other factors that might cause osteoporosis. (See "Epidemiology and causes of osteoporosis"). Calcium-regulating hormones – Disorders of calcium-regulating hormones cause osteoporosis in hyperparathyroidism and rickets or osteomalacia in vitamin D deficiency. These disorders have characteristic biochemical abnormalities of calcium and phosphate balance, and the latter have typical histologic changes that are not found in osteoporosis. (See "Clinical manifestations of primary hyperparathyroidism" and see "Clinical manifestations and etiology of osteomalacia"). Milder degrees of parathyroid hormone (PTH) excess or vitamin D deficiency can contribute to the pathologic changes of osteoporosis. Decreased calcium and vitamin D intake and reduced sun exposure can lead to secondary hyperparathyroidism, which undoubtedly plays a role in age-related bone loss [19]. In one study, for example, women undergoing hip replacement because of hip fracture had lower serum 25-hydroxyvitamin D concentrations than women with or without osteoporosis admitted for elective hip replacement [20]. Hyperparathyroid bone disease is associated with greater loss of cortical than trabecular bone and more preservation of trabecular connections, as compared with osteoporosis. Calcitonin inhibits bone resorption, and it has been thought that calcitonin deficiency could contribute to osteoporosis. However, while bone turnover is decreased in patients given exogenous calcitonin [21], endogenous calcitonin is not an important determinant of osteoporosis. In addition, mice in which the calcitonin gene is deleted have an increase in bone mass [22]. This surprising result suggests that either calcitonin itself or calcitonin gene-related peptide, which is a product of the same gene, has an affect on osteoblasts. Estrogen – The central role of estrogen deficiency in the pathogenesis of osteoporosis in postmenopausal women has been recognized for many years. Estrogen inhibits bone resorption and, after the menopause, estrogen deficiency results in increased bone resorption and rapid bone loss. The rate of loss slows with time after menopause, but the bone loss that continues years after menopause is associated with relatively high levels of markers of bone resorption in many women. Those women aged 70 years or more who continue to produce small amounts of estradiol have a significantly lower risk of hip and spine fractures than those who do not (show figure 4) [23]. Estrogen deficiency may also be important in men as demonstrated by the following observations: • Men with a defect in the estrogen receptor gene or deficiency of aromatase, which converts testosterone to estrogen, have delayed epiphyseal closure and osteoporosis [24,25] • Estrogen therapy leads to epiphyseal closure and an increase in bone mass in men with aromatase deficiency [25] • Serum estrogen concentrations are correlated with bone mass in older men [26,27] • The ability of testosterone therapy to increase bone mass in men is correlated more closely with an increase in serum estrogen than serum testosterone concentrations [28] (See "Overview of osteoporosis in men"). • To study the relative contributions of sex steroids in the regulation of bone resorption and formation (as measured by urinary and serum markers), 59 elderly men (mean age 68 years) received both a GnRH agonist and aromatase inhibitor to eliminate endogenous testosterone and estrogen production, in conjunction with replacement doses of testosterone and estrogen [29]. Subsequent withdrawal of the testosterone, estrogen or both demonstrated that estrogen is the major sex steroid regulating bone resorption, whereas both estrogen and testosterone are important for bone formation. In women, estrogen administration decreases both bone resorption and formation, even 25 to 30 years after menopause [30]. (See "Postmenopausal hormone therapy in the prevention and treatment of osteoporosis"). The decrease in bone formation has been considered secondary to a reduction in the number of resorption cavities where coupled formation normally occurs. Nevertheless, the resorption spaces are filled in, and bone mass usually increases (or at least stops decreasing) with estrogen therapy [31]. The mechanisms by which estrogen regulates bone remodeling are not well understood [32]. It does not inhibit osteoclastic bone resorption in many in vitro systems, but is thought to affect osteoclastogenesis and osteoclast function through its effects on local factors (eg, produced by either bone cells or adjacent marrow cells [33]). Estrogen also decreases the depth of the erosion cavity caused by the osteoclast [34]. (See "Normal skeletal development and regulation of bone formation and resorption"). While there is substantial evidence that estrogen deficiency results in increased bone resorption there may be an additional defect in the bone formation response to increased resorption in estrogen deficiency. Studies in mice have indicated that the anabolic response to mechanical loading is impaired in the absence of estrogen and that this is mediated largely by estrogen receptor alpha [35]. The following observations suggest a pathogenetic role for cytokines and growth factors in the osteoporosis associated with estrogen deficiency; the importance of specific cytokines and growth factors will be discussed in detail below. • An in vitro study showed that estrogen promoted apoptosis of osteoclasts, a change that should reduce osteoclast life span [36]. Estrogen also increased the release of transforming growth factor (TGF)-beta from osteoblasts, and the administration of anti-TGF antibodies inhibited the effect of estrogen on osteoclasts. Thus, the protective effect of estrogen on bone may be mediated in part by TGF-beta. • Tumor necrosis factor-alpha (TNF-a) increases osteoclast recruitment, and its production by peripheral blood monocytes and bone marrow cells is increased after ovariectomy [37]. The possible role of TNF-a in estrogen-deficient bone loss was evaluated in transgenic mice with high serum concentrations of soluble TNF receptor, which blocks the action of TNF-a. Despite ovariectomy, bone density and strength in the transgenic mice were similar to those in control mice [38]. Similarly, in T-cell deficient mice (who therefore cannot produce TNF-a), ovariectomy does not result in increased osteoclast activity and bone loss [39]. These findings suggest that estrogen prevents bone resorption by inhibiting the release of TNF-a. Bone cells and growth plate cartilage contain both the alpha and beta isoforms of the estrogen receptor [40,41]. Estrogen receptors may influence bone cell function by mechanisms that differ from those in the classic target organs, thereby explaining the dissociation between effects on bone and on breast or uterine tissue exerted by selective estrogen receptor modulators tamoxifen and raloxifene. The probable explanation for this dissociation is that the complexes formed by the different compounds with estrogen receptors lead to binding of different trans-activating factors (proteins that interact with DNA and activate or inactivate genes) specific to the target cells [42,43]. (See "Use of selective estrogen receptor modulators in postmenopausal women") Androgens – Androgen deficiency results in bone loss with increased bone turnover similar to that which occurs in estrogen deficiency. In women, the relative importance of androgens (mainly testosterone) and the estrogens (mainly estradiol) derived from androgen metabolism is not known, but androgens may directly stimulate bone formation [44]. Androgen deficiency occurs with aging, and studies in postmenopausal women suggest that low serum androgen concentrations contribute to the bone loss associated with estrogen deficiency [45]. In another report, androgen production, but not estrogen production, was decreased in postmenopausal women with vertebral crush fractures compared with age-matched women with no fractures [46]. Progestins – Bone cells have progesterone receptors, but there is little evidence that progesterone affects bone remodeling in vivo [47]. Cell-culture studies suggest that it may have effects on bone similar to androgen or estrogen [48], but some of these experiments were performed with synthetic progestins that may have androgenic or estrogenic activity. Furthermore, progesterone can interact with glucocorticoid receptors, acting both as an antagonist and weak agonist. Thyroid hormones – Thyroid hormones increase bone resorption and formation; as a result, patients with hyperthyroidism or those treated with excessive doses of thyroxine can have high bone turnover and sometimes low bone density [49]. (See "Bone disease with hyperthyroidism and thyroid hormone therapy"). There is, however, no evidence for a general role of thyroid hormones in the pathogenesis of osteoporosis. Glucocorticoids – Glucocorticoid excess is a common cause of osteoporosis. It differs from primary osteoporosis in that the predominant abnormality is inhibition of bone formation, due to decreases in the replication, migration, differentiation, and life-span of osteoblasts. This is associated with changes in the production of local growth factors, including insulin-like growth factors (IGF) and their binding proteins, and prostaglandins. Increased bone resorption can also occur. This may be due to impaired calcium absorption and secondary hyperparathyroidism, or to decreased estrogen and androgen production. One other factor that may contribute is the underlying inflammatory disorder for which the glucocorticoids are given. (See "Glucocorticoids and osteoporosis: Pathogenesis and clinical features"). Glucocorticoid excess may also play a role in the decreased bone density in patients with major depression, alcoholism, and anorexia nervosa. (See "Glucocorticoids and osteoporosis: Pathogenesis and clinical features"). Growth hormone/insulin-like growth factor – The growth hormone-IGF system is a major determinant of skeletal growth. Growth hormone or IGF-I deficiency, as well as receptor defects, result in dwarfism and diminished bone mass. However, their role in most patients with osteoporosis is probably small [50,51]. There is an age-related decrease in growth hormone secretion and in serum IGF-I and IGF-binding-protein (IGFBP)-3 concentrations in both men and women [52]; in addition, serum IGF-I concentrations are low is some men with idiopathic osteoporosis [53]. IGF-1 is also a local hormone and decreased concentrations in bone have been reported in hip fracture patients [54]. LOCAL CYTOKINES AND PROSTAGLANDINS IN THE PATHOGENESIS OF OSTEOPOROSIS – Bone structure and remodeling are determined by local forces, indicating that there must be important local regulators of bone-cell function. Many local regulators, produced either by adjacent marrow or by bone cells themselves, have been identified. They include cytokines, prostanoids and growth factors. The concept that these factors play a role in the pathogenesis of osteoporosis is supported by animal studies and is consistent with the fact that serum hormone concentrations differ little in patients with osteoporosis and age- and sex-matched control subjects. Many cytokines influence bone-cell function (show table 2). In osteoporosis, the focus has been on an increase in those that stimulate bone resorption, but decreases in cytokines that inhibit resorption, changes in cytokine receptors, or increases in cytokines that inhibit bone formation could also occur. Most of the relevant data are from ovariectomized animals, while data on humans are limited. However, short-term marrow cultures from estrogen-deficient women produce significantly more interleukin (IL)-1, IL-6, TNF-a, and prostaglandin E2 than do similar cultures from estrogen-replete women [37]. The results of studies of peripheral leukocyte cultures and serum concentrations are less consistent but the values were increased in some estrogen-deficient women [55-57]. Interleukin-1 and tumor necrosis factor-alpha – IL-I and TNF-a are potent stimulators of bone resorption and can also inhibit bone formation. Their major sources in bone are probably the marrow cells, particularly macrophages, but IL-1 is also produced by bone cells [58]. Their possible importance in postmenopausal osteoporosis is illustrated by the following observations: • In animals, inhibition of IL-1 and TNF-a by the IL-1 receptor antagonist and a TNF-soluble binding-protein limits the bone loss that occurs after ovariectomy [59]. • Animals lacking IL-1 receptors or transgenic mice with high serum concentrations of soluble TNF receptor, which blocks the action of TNF-a, do not lose bone after ovariectomy [38,60]. • Marrow supernatants from ovariectomized animals stimulate bone resorption by a prostaglandin-dependent mechanism that is inhibited by an IL-1 receptor antagonist [61,62]. • In estrogen-deficient humans IL-1 activity is increased in cultures of peripheral mononuclear cells, and IL-1 mRNA is increased in bone and marrow [37,55-57]. However, concentrations of immunoreactive IL-1 are not consistently increased in supernatants of peripheral blood and marrow cell cultures from patients or animals with estrogen deficiency [63]. One explanation for these results is that IL-1 receptors or binding-proteins rather than IL-1 itself are altered in estrogen deficiency. There are two IL-1 receptors: an activating receptor, IL-1 receptor 1 (IL-1R1), which mediates the action of IL-1; and a "decoy" receptor, IL-1 receptor 2 (IL-1R2), which binds IL-1 but does not mediate a cellular response. The latter receptor can be shed from cells, yet still bind IL-1. The expression of IL-1R2 is decreased by estrogen deficiency and increased by estrogen [64,65]. Interleukin-6 and related cytokines – IL-6 is a major cytokine that is produced by osteoblasts and other cells in the marrow. Stimulators of bone resorption such as PTH, prostaglandin E2 and IL-1 increase IL-6 production in osteoblastic cells [63]. IL-6 stimulates osteoclastogenesis and bone resorption, largely by a prostaglandin-dependent mechanism [66]. Production of IL-6 and its receptors is regulated by sex hormones, and antibodies to IL-6 can block bone loss in orchidectomized animals [67,68]. IL-6 mRNA expression is increased in humans with osteoporosis [69]. In addition, estrogen deficiency is associated with increased production of IL-6 in marrow cultures, but this effect may be indirect because it is blocked when prostaglandin synthesis is inhibited [37]. Serum IL-6 concentrations decrease with age but do not appear to be a marker for osteoporosis, because the concentrations are similar in patients with osteoporosis and age- and sex-matched control subjects [70]. Other cytokines – A number of other cytokines may affect bone function and contribute to bone loss in osteoporosis: • IL-7 stimulates B-cell proliferation and causes bone loss similar to that after ovariectomy [71]. • IL-4 and IL-13 inhibit bone resorption, at least in part by reducing prostaglandin synthesis in bone [72,73]. • Colony-stimulating factor (CSF)-I or macrophage-colony stimulating factor (M-CSF) is essential for the activation of osteoclasts by cytokines. In contrast, IL-18 decreases osteoclastogenesis, probably by increasing granulocyte monocyte-colony stimulating factor (GM-CSF) production and diverting cells away from the osteoclast pathway [74]. Abnormalities of these factors could have a role in osteoporosis, but no studies directly implicate them. Prostaglandins – Prostaglandins are potent regulators of bone cell function [75]. They, particularly prostaglandin E2, increase both bone resorption and formation in both animals and humans. A role for prostaglandins in osteoporosis seems likely, based on the fact that so many of the local and systemic factors that regulate bone metabolism also affect prostaglandin synthesis in bone (show table 2). Endogenous prostaglandins also appear to play a role in the skeletal response to mechanical forces [75,76]. Excessive prostaglandin production might lead to increased bone resorption, while deficient prostaglandin production might impair the bone formation response, both to mechanical loading and remodeling. Prostaglandin production is increased in bones from ovariectomized rats and decreased after estrogen treatment [77]. In addition, the bone resorptive effect of supernatants of cultured marrow cells from ovariectomized mice depends upon their ability to stimulate prostaglandin synthesis [61]. Prostaglandins have also been implicated in the increased bone resorption, but not the reduced bone formation, associated with immobilization [78]. The results of studies on the effect of non-steroidal antiinflammatory drugs (NSAIDs), which inhibit prostaglandin production, are inconsistent. An NSAID-mediated decrease in bone resorption with blunting of bone loss has been reported in animals and humans, but currently available NSAIDs probably cannot be given in sufficient doses to have a sustained effect without serious side effects. New NSAIDs, which are selective for inducible cyclooxygenase (COX-2), may be better tolerated and should be studied for their effects on bone. (See "NSAIDs: Overview of adverse effects"). LOCAL GROWTH FACTORS IN OSTEOPOROSIS – Two major classes of growth factors produced by bone cells could play a role in the pathogenesis of osteoporosis. The IGFs and their binding proteins appear to be most important in maintaining the differentiation and function of osteoblasts [79]. The TGF-beta/bone morphogenic protein family includes potent mitogens for bone cell precursors, but some members of the family are most important in embryonic differentiation [80,81]. As noted above, polymorphisms in the BMP-2 gene have been linked to osteoporosis [14]. Insulin-like growth factor-I – The age-related decrease in serum IGF-I concentrations is paralleled by an age-related decrease in the skeletal content of both IGF-I and IGF-II [54]. Estrogen's effects on this system are inconsistent; it stimulates IGF production in cultured bone cells, but the IGF content of bone of ovariectomized rats is normal [82]. IGFs may play their largest role in the pathogenesis of idiopathic low-turnover osteoporosis in men and premenopausal women [83]. Transforming growth factor-beta – TGF-beta is abundant in bone. It can inhibit bone resorption and stimulate bone formation, and could play a role as a coupling factor. As an example, TGF-beta might be released from bone cells and activated during resorption, diminishing the activity of osteoclasts by accelerating apoptosis of these cells [36]. TGF-beta might then initiate bone formation by stimulating replication and differentiation of osteoblast precursors. Part of the protective effect of estrogen against bone loss could be mediated by TGF-beta-induced apoptosis of osteoclasts [36]. Based upon this role, a deficiency of TGF-beta could cause osteoporosis. Compatible with this hypothesis is the observation that ovariectomy in rodents is associated with a decrease in the TGF-beta mRNA and protein content in bone matrix [82,84]. However, animals in which TGF-beta is overexpressed in bone paradoxically have severe osteoporosis [85]. Thus, the role of TGF-beta in the pathogenesis of osteoporosis remains uncertain. A particular polymorphism in TGF-beta-1 is more prevalent in women with osteoporosis and is associated with low bone mass and increased bone turnover in both osteoporotic and normal women [86]. PTH-related protein – PTH-related protein (PTHrP) is produced by bone and cartilage cells. It has an important regulatory function in the development of bone and cartilage, and may also have a local regulatory function in adult bone. PTHrP, which is secreted by the lactating mammary gland, may play a role in the increased rate of bone resorption and rapid bone loss that occurs in lactating women. This was demonstrated in a mouse model in which PTHrP was eliminated from mammary epithelial cells during late pregnancy and lactation, resulting in a decrease in bone turnover and bone loss [87]. Fibroblast growth factor – Fibroblast growth factor is produced by bone cells as well as other connective tissue cells, and its production is regulated by PTH, prostaglandin, E2 and TGF-beta. It decreases collagen synthesis in vitro, but can stimulate bone formation in vivo [88,89]. CONCLUSIONS – The complex disorder that we call osteoporosis has many subtypes with different pathogenetic factors, and these factors may change with time, thereby changing the pathophysiology. Thus, an initial increase in bone resorption may be followed by a failure of bone formation in the most severe and progressive cases. The preceding summary of the factors that influence bone metabolism can only suggest their possible pathogenic roles. Genetic analyses and evaluation of local factors, by in situ hybridization and immunocytochemistry, and new imaging approaches to determine bone structure should help us better understand osteoporosis in the future.
 [1]. • Low bone mass is a characteristic finding in osteoporosis. Figure 1 shows normal bone in panel A, and progressively more osteoporotic bone in panels B, C and D (show figure 1). The bone that is present is normally mineralized, which distinguishes osteoporosis from osteomalacia. • There is disruption of the normal architecture, as illustrated in Figure 1, panel D (show figure 1). There are fewer bony spicules in osteoporotic bone and they are thinner than normal; in addition, there are horizontal struts that do not join up to any other structure, and thereby provide no structural support. This microarchitectural disruption undermines the structural integrity of the bone, and leads to the major clinical features of osteoporosis: skeletal fragility and an increase in fracture risk [1]. The mechanisms of the microarchitectural disruption are not clear. Increased remodeling itself may cause structural weakening, which may account for the independent association of high bone turnover with fracture risk (show figure 2) [2]. Other possible factors include: • Microfractures and fatigue damage. • The development of perforations and discontinuities in trabecular bone, as well as a relatively excessive loss of horizontal trabeculae (show figure 1) • Macroarchitecture may play a role; as an example, increased length of the femoral neck appears to increase the risk of hip fracture. • Posture, muscle strength and the frequency and type of falls affect fracture frequency and site. Decreased bone mass can occur because peak bone mass is low, bone resorption is excessive, or bone formation during remodeling is decreased. All three processes are likely to contribute, in varying degrees, to osteoporosis in individual patients. Their relative contribution to fracture risk is not known, but it seems likely that increased bone resorption has the greatest impact [3,4]. Age- and menopause-related bone loss are clearly important pathogenetic factors, but their expression must vary because there are wide variations in the amount of bone and the amount of porosity of bone in older persons of the same age (show figure 3) [5]. This topic will focus on the determinants of bone mass and remodeling that are likely to be important in the pathogenesis of decreased bone density. The epidemiology and causes of osteoporosis and the factors that normally regulate bone formation and resorption are discussed separately. (See Epidemiology and causes of osteoporosis and see Normal skeletal development and regulation of bone formation and resorption ). DETERMINANTS OF PEAK BONE MASS – Based upon twin studies, genetic determinants account for 40 to 80 percent of the differences in peak bone mass. Skeletal structure and bone turnover are probably also genetically determined, but environmental factors affect bone growth during childhood and adolescence. Thus, increasing calcium intake and physical activity have a small positive affect on peak bone mineral density, moreover the increment in peak bone mass that occurs due to these lifestyle factors may have a substantial affect on the later incidence of fragility fractures [6]. As examples, calcium-enriched foods or supplements (approximately 1600 mg/day) promoted accrual of bone in children [7,8]. The effect lasted three to five years after the end of the supplementation in one, but not another study [9,10] (See Calcium requirements in adolescents ). There may be important interactions between genetic and environmental factors. As an example, the differences in bone mass attributed to different alleles of the vitamin D receptor may be dependent upon differences in calcium intake [11]. Many genes have been examined for their possible role in the pathogenesis of osteoporosis [12,13]. • Different alleles of the gene for the vitamin D receptor are associated with small differences in bone mass [11]. However, the differences are not consistent among studies, and there is little evidence for a general association between vitamin D receptor alleles and osteoporosis [12,13]. • Increased frequency of osteoporosis has been reported in patients with a particular polymorphism of an Sp-1 cleavage site in the first intron of the collagen gene [12,13]. • Other genes, including those for the estrogen receptor, transforming growth factor-beta, and apolipoprotein E, have also been implicated in osteoporosis. • A report based upon an extensive linkage analysis suggested that polymorphisms in the bone morphogenetic protein-2 (BMP-2) gene were associated with an increased risk of fracture and low BMD in both pre- and postmenopausal women [14]. • An activating mutation of the gene for a low-density-lipoprotein (LDL) receptor-related protein 5 (LRP-5) was associated with high bone mass as an autosomal dominant in several families [15,16]. Transgenic mice carrying the activating mutant have increased bone mass and strength. Deletion of the LRP-5 gene causes an unusual autosomal recessive disorder, osteoporosis-pseudoglioma syndrome, in which bone mass is markedly reduced [17]. Whether alterations in the expression or activity of this gene and related signal transduction molecules are important in the pathogenesis of osteoporosis is not known. Not enough is known about the genetics of osteoporosis to affect treatment. The selection of patients for diagnostic work-up and preventive therapy could be improved if there were good genetic markers for risk. At least, some of the variability in the clinical course of osteoporosis might be explained. MECHANISMS OF BONE LOSS IN OSTEOPOROSIS – Bone loss can occur because bone resorption is increased or bone formation is decreased. There is considerable evidence that osteoporosis is associated with increased bone resorption, consistent with the morphologic pattern of trabecular bone loss and increased cortical porosity. However, bone biopsies in patients with osteoporosis do not clearly show an increase in actively resorbing surfaces [18]. Therefore, as compared with bone resorption, there must be a relative decrease in bone formation to account for bone loss. An increase in activation frequency, ie, in the number of bone-resorbing sites, leads to a transient decrease in bone mass. In normal subjects, there is a compensatory increase in bone formation, leading to restoration of bone mass. However, if the increase in resorption results in perforations of trabecular plates or discontinuities of trabecular struts, the template for new bone formation is lost and the decrease in bone mass is irreversible. The term high turnover osteoporosis has been used to describe patients in whom excessive bone resorption predominates, while low turnover has been applied to patients in whom defective formation predominates. However, the relative contributions of increased resorption and decreased formation probably represent a continuum, and this relation may change during the course of the disease. Biopsies of patients with advanced osteoporosis usually show decreased osteoblastic activity, but this may be an end stage of a process that began with excessive resorption. ROLE OF SYSTEMIC HORMONES IN OSTEOPOROSIS – Age- and menopause-related changes in the production of many hormones have been identified, but there are no differences in the serum concentrations of any hormone in patients with osteoporosis and suitably matched control subjects. Nevertheless, the changes in calcium-regulating hormones, sex hormones, and growth-regulating hormones that occur with age probably contribute to osteoporosis or at least result in increased susceptibility to other factors that might cause osteoporosis. (See Epidemiology and causes of osteoporosis ). Calcium-regulating hormones – Disorders of calcium-regulating hormones cause osteoporosis in hyperparathyroidism and rickets or osteomalacia in vitamin D deficiency. These disorders have characteristic biochemical abnormalities of calcium and phosphate balance, and the latter have typical histologic changes that are not found in osteoporosis. (See Clinical manifestations of primary hyperparathyroidism and see Clinical manifestations and etiology of osteomalacia ). Milder degrees of parathyroid hormone (PTH) excess or vitamin D deficiency can contribute to the pathologic changes of osteoporosis. Decreased calcium and vitamin D intake and reduced sun exposure can lead to secondary hyperparathyroidism, which undoubtedly plays a role in age-related bone loss [19]. In one study, for example, women undergoing hip replacement because of hip fracture had lower serum 25-hydroxyvitamin D concentrations than women with or without osteoporosis admitted for elective hip replacement [20]. Hyperparathyroid bone disease is associated with greater loss of cortical than trabecular bone and more preservation of trabecular connections, as compared with osteoporosis. Calcitonin inhibits bone resorption, and it has been thought that calcitonin deficiency could contribute to osteoporosis. However, while bone turnover is decreased in patients given exogenous calcitonin [21], endogenous calcitonin is not an important determinant of osteoporosis. In addition, mice in which the calcitonin gene is deleted have an increase in bone mass [22]. This surprising result suggests that either calcitonin itself or calcitonin gene-related peptide, which is a product of the same gene, has an affect on osteoblasts. Estrogen – The central role of estrogen deficiency in the pathogenesis of osteoporosis in postmenopausal women has been recognized for many years. Estrogen inhibits bone resorption and, after the menopause, estrogen deficiency results in increased bone resorption and rapid bone loss. The rate of loss slows with time after menopause, but the bone loss that continues years after menopause is associated with relatively high levels of markers of bone resorption in many women. Those women aged 70 years or more who continue to produce small amounts of estradiol have a significantly lower risk of hip and spine fractures than those who do not (show figure 4) [23]. Estrogen deficiency may also be important in men as demonstrated by the following observations: • Men with a defect in the estrogen receptor gene or deficiency of aromatase, which converts testosterone to estrogen, have delayed epiphyseal closure and osteoporosis [24,25] • Estrogen therapy leads to epiphyseal closure and an increase in bone mass in men with aromatase deficiency [25] • Serum estrogen concentrations are correlated with bone mass in older men [26,27] • The ability of testosterone therapy to increase bone mass in men is correlated more closely with an increase in serum estrogen than serum testosterone concentrations [28] (See Overview of osteoporosis in men ). • To study the relative contributions of sex steroids in the regulation of bone resorption and formation (as measured by urinary and serum markers), 59 elderly men (mean age 68 years) received both a GnRH agonist and aromatase inhibitor to eliminate endogenous testosterone and estrogen production, in conjunction with replacement doses of testosterone and estrogen [29]. Subsequent withdrawal of the testosterone, estrogen or both demonstrated that estrogen is the major sex steroid regulating bone resorption, whereas both estrogen and testosterone are important for bone formation. In women, estrogen administration decreases both bone resorption and formation, even 25 to 30 years after menopause [30]. (See Postmenopausal hormone therapy in the prevention and treatment of osteoporosis ). The decrease in bone formation has been considered secondary to a reduction in the number of resorption cavities where coupled formation normally occurs. Nevertheless, the resorption spaces are filled in, and bone mass usually increases (or at least stops decreasing) with estrogen therapy [31]. The mechanisms by which estrogen regulates bone remodeling are not well understood [32]. It does not inhibit osteoclastic bone resorption in many in vitro systems, but is thought to affect osteoclastogenesis and osteoclast function through its effects on local factors (eg, produced by either bone cells or adjacent marrow cells [33]). Estrogen also decreases the depth of the erosion cavity caused by the osteoclast [34]. (See Normal skeletal development and regulation of bone formation and resorption ). While there is substantial evidence that estrogen deficiency results in increased bone resorption there may be an additional defect in the bone formation response to increased resorption in estrogen deficiency. Studies in mice have indicated that the anabolic response to mechanical loading is impaired in the absence of estrogen and that this is mediated largely by estrogen receptor alpha [35]. The following observations suggest a pathogenetic role for cytokines and growth factors in the osteoporosis associated with estrogen deficiency; the importance of specific cytokines and growth factors will be discussed in detail below. • An in vitro study showed that estrogen promoted apoptosis of osteoclasts, a change that should reduce osteoclast life span [36]. Estrogen also increased the release of transforming growth factor (TGF)-beta from osteoblasts, and the administration of anti-TGF antibodies inhibited the effect of estrogen on osteoclasts. Thus, the protective effect of estrogen on bone may be mediated in part by TGF-beta. • Tumor necrosis factor-alpha (TNF-a) increases osteoclast recruitment, and its production by peripheral blood monocytes and bone marrow cells is increased after ovariectomy [37]. The possible role of TNF-a in estrogen-deficient bone loss was evaluated in transgenic mice with high serum concentrations of soluble TNF receptor, which blocks the action of TNF-a. Despite ovariectomy, bone density and strength in the transgenic mice were similar to those in control mice [38]. Similarly, in T-cell deficient mice (who therefore cannot produce TNF-a), ovariectomy does not result in increased osteoclast activity and bone loss [39]. These findings suggest that estrogen prevents bone resorption by inhibiting the release of TNF-a. Bone cells and growth plate cartilage contain both the alpha and beta isoforms of the estrogen receptor [40,41]. Estrogen receptors may influence bone cell function by mechanisms that differ from those in the classic target organs, thereby explaining the dissociation between effects on bone and on breast or uterine tissue exerted by selective estrogen receptor modulators tamoxifen and raloxifene. The probable explanation for this dissociation is that the complexes formed by the different compounds with estrogen receptors lead to binding of different trans-activating factors (proteins that interact with DNA and activate or inactivate genes) specific to the target cells [42,43]. (See Use of selective estrogen receptor modulators in postmenopausal women ) Androgens – Androgen deficiency results in bone loss with increased bone turnover similar to that which occurs in estrogen deficiency. In women, the relative importance of androgens (mainly testosterone) and the estrogens (mainly estradiol) derived from androgen metabolism is not known, but androgens may directly stimulate bone formation [44]. Androgen deficiency occurs with aging, and studies in postmenopausal women suggest that low serum androgen concentrations contribute to the bone loss associated with estrogen deficiency [45]. In another report, androgen production, but not estrogen production, was decreased in postmenopausal women with vertebral crush fractures compared with age-matched women with no fractures [46]. Progestins – Bone cells have progesterone receptors, but there is little evidence that progesterone affects bone remodeling in vivo [47]. Cell-culture studies suggest that it may have effects on bone similar to androgen or estrogen [48], but some of these experiments were performed with synthetic progestins that may have androgenic or estrogenic activity. Furthermore, progesterone can interact with glucocorticoid receptors, acting both as an antagonist and weak agonist. Thyroid hormones – Thyroid hormones increase bone resorption and formation; as a result, patients with hyperthyroidism or those treated with excessive doses of thyroxine can have high bone turnover and sometimes low bone density [49]. (See Bone disease with hyperthyroidism and thyroid hormone therapy ). There is, however, no evidence for a general role of thyroid hormones in the pathogenesis of osteoporosis. Glucocorticoids – Glucocorticoid excess is a common cause of osteoporosis. It differs from primary osteoporosis in that the predominant abnormality is inhibition of bone formation, due to decreases in the replication, migration, differentiation, and life-span of osteoblasts. This is associated with changes in the production of local growth factors, including insulin-like growth factors (IGF) and their binding proteins, and prostaglandins. Increased bone resorption can also occur. This may be due to impaired calcium absorption and secondary hyperparathyroidism, or to decreased estrogen and androgen production. One other factor that may contribute is the underlying inflammatory disorder for which the glucocorticoids are given. (See Glucocorticoids and osteoporosis: Pathogenesis and clinical features ). Glucocorticoid excess may also play a role in the decreased bone density in patients with major depression, alcoholism, and anorexia nervosa. (See Glucocorticoids and osteoporosis: Pathogenesis and clinical features ). Growth hormone/insulin-like growth factor – The growth hormone-IGF system is a major determinant of skeletal growth. Growth hormone or IGF-I deficiency, as well as receptor defects, result in dwarfism and diminished bone mass. However, their role in most patients with osteoporosis is probably small [50,51]. There is an age-related decrease in growth hormone secretion and in serum IGF-I and IGF-binding-protein (IGFBP)-3 concentrations in both men and women [52]; in addition, serum IGF-I concentrations are low is some men with idiopathic osteoporosis [53]. IGF-1 is also a local hormone and decreased concentrations in bone have been reported in hip fracture patients [54]. LOCAL CYTOKINES AND PROSTAGLANDINS IN THE PATHOGENESIS OF OSTEOPOROSIS – Bone structure and remodeling are determined by local forces, indicating that there must be important local regulators of bone-cell function. Many local regulators, produced either by adjacent marrow or by bone cells themselves, have been identified. They include cytokines, prostanoids and growth factors. The concept that these factors play a role in the pathogenesis of osteoporosis is supported by animal studies and is consistent with the fact that serum hormone concentrations differ little in patients with osteoporosis and age- and sex-matched control subjects. Many cytokines influence bone-cell function (show table 2). In osteoporosis, the focus has been on an increase in those that stimulate bone resorption, but decreases in cytokines that inhibit resorption, changes in cytokine receptors, or increases in cytokines that inhibit bone formation could also occur. Most of the relevant data are from ovariectomized animals, while data on humans are limited. However, short-term marrow cultures from estrogen-deficient women produce significantly more interleukin (IL)-1, IL-6, TNF-a, and prostaglandin E2 than do similar cultures from estrogen-replete women [37]. The results of studies of peripheral leukocyte cultures and serum concentrations are less consistent but the values were increased in some estrogen-deficient women [55-57]. Interleukin-1 and tumor necrosis factor-alpha – IL-I and TNF-a are potent stimulators of bone resorption and can also inhibit bone formation. Their major sources in bone are probably the marrow cells, particularly macrophages, but IL-1 is also produced by bone cells [58]. Their possible importance in postmenopausal osteoporosis is illustrated by the following observations: • In animals, inhibition of IL-1 and TNF-a by the IL-1 receptor antagonist and a TNF-soluble binding-protein limits the bone loss that occurs after ovariectomy [59]. • Animals lacking IL-1 receptors or transgenic mice with high serum concentrations of soluble TNF receptor, which blocks the action of TNF-a, do not lose bone after ovariectomy [38,60]. • Marrow supernatants from ovariectomized animals stimulate bone resorption by a prostaglandin-dependent mechanism that is inhibited by an IL-1 receptor antagonist [61,62]. • In estrogen-deficient humans IL-1 activity is increased in cultures of peripheral mononuclear cells, and IL-1 mRNA is increased in bone and marrow [37,55-57]. However, concentrations of immunoreactive IL-1 are not consistently increased in supernatants of peripheral blood and marrow cell cultures from patients or animals with estrogen deficiency [63]. One explanation for these results is that IL-1 receptors or binding-proteins rather than IL-1 itself are altered in estrogen deficiency. There are two IL-1 receptors: an activating receptor, IL-1 receptor 1 (IL-1R1), which mediates the action of IL-1; and a decoy receptor, IL-1 receptor 2 (IL-1R2), which binds IL-1 but does not mediate a cellular response. The latter receptor can be shed from cells, yet still bind IL-1. The expression of IL-1R2 is decreased by estrogen deficiency and increased by estrogen [64,65]. Interleukin-6 and related cytokines – IL-6 is a major cytokine that is produced by osteoblasts and other cells in the marrow. Stimulators of bone resorption such as PTH, prostaglandin E2 and IL-1 increase IL-6 production in osteoblastic cells [63]. IL-6 stimulates osteoclastogenesis and bone resorption, largely by a prostaglandin-dependent mechanism [66]. Production of IL-6 and its receptors is regulated by sex hormones, and antibodies to IL-6 can block bone loss in orchidectomized animals [67,68]. IL-6 mRNA expression is increased in humans with osteoporosis [69]. In addition, estrogen deficiency is associated with increased production of IL-6 in marrow cultures, but this effect may be indirect because it is blocked when prostaglandin synthesis is inhibited [37]. Serum IL-6 concentrations decrease with age but do not appear to be a marker for osteoporosis, because the concentrations are similar in patients with osteoporosis and age- and sex-matched control subjects [70]. Other cytokines – A number of other cytokines may affect bone function and contribute to bone loss in osteoporosis: • IL-7 stimulates B-cell proliferation and causes bone loss similar to that after ovariectomy [71]. • IL-4 and IL-13 inhibit bone resorption, at least in part by reducing prostaglandin synthesis in bone [72,73]. • Colony-stimulating factor (CSF)-I or macrophage-colony stimulating factor (M-CSF) is essential for the activation of osteoclasts by cytokines. In contrast, IL-18 decreases osteoclastogenesis, probably by increasing granulocyte monocyte-colony stimulating factor (GM-CSF) production and diverting cells away from the osteoclast pathway [74]. Abnormalities of these factors could have a role in osteoporosis, but no studies directly implicate them. Prostaglandins – Prostaglandins are potent regulators of bone cell function [75]. They, particularly prostaglandin E2, increase both bone resorption and formation in both animals and humans. A role for prostaglandins in osteoporosis seems likely, based on the fact that so many of the local and systemic factors that regulate bone metabolism also affect prostaglandin synthesis in bone (show table 2). Endogenous prostaglandins also appear to play a role in the skeletal response to mechanical forces [75,76]. Excessive prostaglandin production might lead to increased bone resorption, while deficient prostaglandin production might impair the bone formation response, both to mechanical loading and remodeling. Prostaglandin production is increased in bones from ovariectomized rats and decreased after estrogen treatment [77]. In addition, the bone resorptive effect of supernatants of cultured marrow cells from ovariectomized mice depends upon their ability to stimulate prostaglandin synthesis [61]. Prostaglandins have also been implicated in the increased bone resorption, but not the reduced bone formation, associated with immobilization [78]. The results of studies on the effect of non-steroidal antiinflammatory drugs (NSAIDs), which inhibit prostaglandin production, are inconsistent. An NSAID-mediated decrease in bone resorption with blunting of bone loss has been reported in animals and humans, but currently available NSAIDs probably cannot be given in sufficient doses to have a sustained effect without serious side effects. New NSAIDs, which are selective for inducible cyclooxygenase (COX-2), may be better tolerated and should be studied for their effects on bone. (See NSAIDs: Overview of adverse effects ). LOCAL GROWTH FACTORS IN OSTEOPOROSIS – Two major classes of growth factors produced by bone cells could play a role in the pathogenesis of osteoporosis. The IGFs and their binding proteins appear to be most important in maintaining the differentiation and function of osteoblasts [79]. The TGF-beta/bone morphogenic protein family includes potent mitogens for bone cell precursors, but some members of the family are most important in embryonic differentiation [80,81]. As noted above, polymorphisms in the BMP-2 gene have been linked to osteoporosis [14]. Insulin-like growth factor-I – The age-related decrease in serum IGF-I concentrations is paralleled by an age-related decrease in the skeletal content of both IGF-I and IGF-II [54]. Estrogen s effects on this system are inconsistent; it stimulates IGF production in cultured bone cells, but the IGF content of bone of ovariectomized rats is normal [82]. IGFs may play their largest role in the pathogenesis of idiopathic low-turnover osteoporosis in men and premenopausal women [83]. Transforming growth factor-beta – TGF-beta is abundant in bone. It can inhibit bone resorption and stimulate bone formation, and could play a role as a coupling factor. As an example, TGF-beta might be released from bone cells and activated during resorption, diminishing the activity of osteoclasts by accelerating apoptosis of these cells [36]. TGF-beta might then initiate bone formation by stimulating replication and differentiation of osteoblast precursors. Part of the protective effect of estrogen against bone loss could be mediated by TGF-beta-induced apoptosis of osteoclasts [36]. Based upon this role, a deficiency of TGF-beta could cause osteoporosis. Compatible with this hypothesis is the observation that ovariectomy in rodents is associated with a decrease in the TGF-beta mRNA and protein content in bone matrix [82,84]. However, animals in which TGF-beta is overexpressed in bone paradoxically have severe osteoporosis [85]. Thus, the role of TGF-beta in the pathogenesis of osteoporosis remains uncertain. A particular polymorphism in TGF-beta-1 is more prevalent in women with osteoporosis and is associated with low bone mass and increased bone turnover in both osteoporotic and normal women [86]. PTH-related protein – PTH-related protein (PTHrP) is produced by bone and cartilage cells. It has an important regulatory function in the development of bone and cartilage, and may also have a local regulatory function in adult bone. PTHrP, which is secreted by the lactating mammary gland, may play a role in the increased rate of bone resorption and rapid bone loss that occurs in lactating women. This was demonstrated in a mouse model in which PTHrP was eliminated from mammary epithelial cells during late pregnancy and lactation, resulting in a decrease in bone turnover and bone loss [87]. Fibroblast growth factor – Fibroblast growth factor is produced by bone cells as well as other connective tissue cells, and its production is regulated by PTH, prostaglandin, E2 and TGF-beta. It decreases collagen synthesis in vitro, but can stimulate bone formation in vivo [88,89]. CONCLUSIONS – The complex disorder that we call osteoporosis has many subtypes with different pathogenetic factors, and these factors may change with time, thereby changing the pathophysiology. Thus, an initial increase in bone resorption may be followed by a failure of bone formation in the most severe and progressive cases. The preceding summary of the factors that influence bone metabolism can only suggest their possible pathogenic roles. Genetic analyses and evaluation of local factors, by in situ hybridization and immunocytochemistry, and new imaging approaches to determine bone structure should help us better understand osteoporosis in the future.")
9
Parathormon Paratiroid bezlerinden salgılanır.
84 amino asitli bir polipeptiddir. Plazma düzeyi 1 ng/mL Etkileri D vitaminine benzer; ondan farklı olarak böbreklerden fosfat atılımını azaltmayıp, tersine artırır.

11
Parathormon Preparatları
Para-thor-mone 500 U/5 mL viyal, 5 viyal, (Lilly, İngiltere) Doz: günde 2 kez Ü (i.m. veya s.k.), birkaç gün verilir. PTH preparatları alerjik reaksiyonlara neden olabilir. (Kasaplık hayvanlardan elde edildiği için) Uzun süre kullanıldığında antikor gelişmesi nedeniyle etkisine karşı tolerans gelişir. Paratiroid hormonun rekombinan bir fragmanı olan teriparatid menapoz sonrası osteoporoz tedavisinde kullanılmak üzere üretilmiştir. Teriparatid FORSTEO 750 micg/3 mL enj. kalemi 772,16 YTL
 Doz: günde 2 kez Ü (i.m. veya s.k.), birkaç gün verilir. PTH preparatları alerjik reaksiyonlara neden olabilir. (Kasaplık hayvanlardan elde edildiği için) Uzun süre kullanıldığında antikor gelişmesi nedeniyle etkisine karşı tolerans gelişir. Paratiroid hormonun rekombinan bir fragmanı olan teriparatid menapoz sonrası osteoporoz tedavisinde kullanılmak üzere üretilmiştir. Teriparatid. FORSTEO 750 micg/3 mL enj. kalemi 772,16 YTL.")
12
Hipoparatiroidizm En önemli belirti hipokalsemidir. Buna bağlı olarak tetani gelişir. Özellikle tiroid cerrahisi sırasında paratiroid bezlerinin de yanlışlıkla çıkartılması nedeniyle oluşur. TEDAVİ Kalsiyum tuzları Kalsitriol Yüksek doz D vitamini Rutin tedavide PTH kullanılmaz Psödohipoparatiroidizm Paratiroid bezleri ve salgısı normaldir. Periferik direnç vardır. Antireseptör antikorlar Gs düzeyinde bir bozukluk (bazı hastalarda)
")
13
Hiperparatiroidizm Tümör (primer): Kronik hipokalsemi (sekonder):
hiperkalsemi hipofosfatemi alkalin fosfataz hiperkalsiüri (ürolitiazis sıklığı ) osteoporoz Kronik hipokalsemi (sekonder): Kronik böbrek hastalıkları D vitamini eksikliği Bazı kronik hastalık halleri hipokalsemi paratiroid hiperplazisi
 osteoporoz. Kronik hipokalsemi (sekonder): Kronik böbrek hastalıkları. D vitamini eksikliği. Bazı kronik hastalık halleri. hipokalsemi paratiroid hiperplazisi.")
14
Kolekalsiferol (D3 vit.)
D Vitamini Kaynakları bakımından farklı, fakat yapı ve oluşumları bakımından birbirine benzeyen 2 türlü D vitamini vardır. Kalsiferol (D2 vit.) Bitkiler içinde bulunan, bir ön vitamin olan ergosterol şeklinde alınır; ciltte toplanır. Kolekalsiferol (D3 vit.) Vücutta sentezlenir. Bu nedenle gerçek bir vitamin değil, bir hormon prekürsörüdür. Hayvansal kaynaklı besinler içinde de alınabilir (balıklar; özellikle sardalya eti ve balık yağı). Süt D vitamini içeriği yönünden zengin değildir.
 Bitkiler içinde bulunan, bir ön vitamin olan ergosterol şeklinde alınır; ciltte toplanır. Kolekalsiferol (D3 vit.) Vücutta sentezlenir. Bu nedenle gerçek bir vitamin değil, bir hormon prekürsörüdür. Hayvansal kaynaklı besinler içinde de alınabilir (balıklar; özellikle sardalya eti ve balık yağı). Süt D vitamini içeriği yönünden zengin değildir.")
15
Vitamin D Nuclear Receptor Three-dimensional model of the ligand binding domain (LBD) of the nuclear receptor (VDR) for the steroid hormone, 1a,25(OH)2-vitamin D3 [1a,25(OH)2D3], based on the atomic coordinates of the nuclear receptor for thyroid hormone. The left panel illustrates the twelve a-helices of the protein (presented as ribbons) and four b-strands that collectively define the LBD of the VDR. Each helix has its own unique color. The right panel depicts 1a,25(OH)2D3 (blue colored molecule) beginning to enter the VDR LBD, which is a CP space-filling representation; the color-coding of the 12 helices of the VDR is the same as in the left panel.
![Vitamin D Nuclear Receptor Three-dimensional model of the ligand binding domain (LBD) of the nuclear receptor (VDR) for the steroid hormone, 1a,25(OH)2-vitamin D3 [1a,25(OH)2D3], based on the atomic coordinates of the nuclear receptor for thyroid hormone. The left panel illustrates the twelve a-helices of the protein (presented as ribbons) and four b-strands that collectively define the LBD of the VDR. Each helix has its own unique color.](http://slideplayer.biz.tr/slide/2595228/9/images/15/Vitamin+D+Nuclear+Receptor+Three-dimensional+model+of+the+ligand+binding+domain+%28LBD%29+of+the+nuclear+receptor+%28VDR%29+for+the+steroid+hormone%2C+1a%2C25%28OH%292-vitamin+D3+%5B1a%2C25%28OH%292D3%5D%2C+based+on+the+atomic+coordinates+of+the+nuclear+receptor+for+thyroid+hormone.+The+left+panel+illustrates+the+twelve+a-helices+of+the+protein+%28presented+as+ribbons%29+and+four+b-strands+that+collectively+define+the+LBD+of+the+VDR.+Each+helix+has+its+own+unique+color..jpg "The right panel depicts 1a,25(OH)2D3 (blue colored molecule) beginning to enter the VDR LBD, which is a CP space-filling representation; the color-coding of the 12 helices of the VDR is the same as in the left panel.")
16
Günlük D Vitamini Gereksinimi
< 18 yaş : 400 Ü (10 g kolekalsiferol eşdeğeri) 19-23 yaş : 300 Ü > 23 yaş : 200 Ü Gebelik ve laktasyonda günlük gereksinim 200 Ü artar.
 yaş : 300 Ü. > 23 yaş : 200 Ü. Gebelik ve laktasyonda günlük gereksinim 200 Ü artar.")
17
D Vitamini Metabolizması
Absorbsiyon İnce barsaktan absorbe edilirler. D3 daha hızlı ve daha fazla absorbe edilir. Absorbsiyonları safra asitlerine gereksinim duyar. Dağılım D vitamini bağlayan proteine bağlanarak taşınırlar. Karaciğer ve yağ dokusunda depolanırlar.

18
D Vitaminlerinin Biyoaktivasyonu

20
D Vitamini Metabolizması
Eliminasyon Karaciğerde hidroksillenme ve konjugasyon mekanizmaları ile inaktive edilirler (karaciğer mikrozomal enzimleri bu olayda kısmen rol oynar). Metabolitlerin büyük bir kısmı safra içinde atılırlar ve enterohepatik dolanıma girerler. Fenitoin ve fenobarbital enzim indüksiyonu yaparak inaktivasyonu hızlandırırlar ve uzun süreli kullanılmaları ile D vitamini eksikliğine yol açabilirler. İzoniazid D vitamininin aktif hidroksilli türevlerine dönüşmesini engeller; izoniazid ile birlikte profilaktik olarak D vitamini verilmelidir.
. Metabolitlerin büyük bir kısmı safra içinde atılırlar ve enterohepatik dolanıma girerler. Fenitoin ve fenobarbital enzim indüksiyonu yaparak inaktivasyonu hızlandırırlar ve uzun süreli kullanılmaları ile D vitamini eksikliğine yol açabilirler. İzoniazid D vitamininin aktif hidroksilli türevlerine dönüşmesini engeller; izoniazid ile birlikte profilaktik olarak D vitamini verilmelidir.")
21
D vitamini eksikliği Çocuklarda raşitizm (rickets)
Erişkinlerde osteomalasi CAUSES OF VITAMINE D DEFICIENCY AND RESISTANCE INTRODUCTION – Vitamin D has a variety of actions on calcium, phosphate, and bone metabolism. By increasing intestinal calcium and phosphate reabsorption and increasing the effect of parathyroid hormone (PTH) on bone, vitamin D has the net effect of increasing the serum calcium and phosphate concentrations (show figure 1). Vitamin D deficiency or resistance interferes with these processes, causing hypocalcemia and hypophosphatemia. The latter is usually more prominent, since hypocalcemia stimulates the release of PTH. The secondary hyperparathyroidism, via its actions on bone and the kidney, partially corrects the hypocalcemia but worsens the hypophosphatemia by increasing urinary phosphate excretion. (See "Chapter 6F: Hormonal regulation of calcium and phosphate balance"). This topic will review the major causes of vitamin D deficiency or resistance. The major causes of the clinical manifestations of this problem – hypophosphatemia and hypocalcemia – are discussed separately. (See "Causes of hypophosphatemia" and see "Etiology of hypocalcemia in adults"). ETIOLOGY – Vitamin D deficiency can occur as a result of decreased intake or absorption, reduced sun exposure, increased hepatic catabolism, decreased endogenous synthesis (via 25-hydroxylation in the liver and subsequent 1-hydroxylation in the kidney), or end-organ resistance (show table 1) [1]. (See "Metabolism of vitamin D"). Reduced vitamin D intake or production in skin – In the United States, most vitamin D is derived from foods that are rich in the vitamin (fatty fishes) or fortified with the vitamin (milk and related products and cereals). The remainder is synthesized in the skin from 7-dehydrocholesterol under the influence of ultraviolet light (show figure 1). Vitamin D deficiency can therefore occur in people who live without sun exposure (including those whose skin is constantly protected from the sun) and whose dietary intake is low, which is most common in countries distant from the equator in which foods are not fortified with vitamin D [2]. Cutaneous vitamin D production and vitamin D stores decline with age [3]. This change is most prominent in the winter. In temperate areas such as Boston and Edmonton, for example, cutaneous production of vitamin D virtually ceases in winter, especially in the elderly [4,5]. It may also be more common in Asian Indian immigrants to the United States who may have vitamin D deficiency, even with adequate sun exposure [6]. In patients with a history of extensive burn injuries, vitamin D synthesis in skin is below normal, even with sun exposure [7]. Thus, these patients should receive vitamin D supplementation. In addition to reduced endogenous production, vitamin D intake is often low in older subjects. It has been estimated that approximately one-half of elderly women consume less than 137 IU/day of vitamin D, and nearly one-quarter consume less than 65 IU/day (recommended intake 400 IU/day for people 51 to 70 years old and 600 IU/day for people 71 years old and older) [5]. The net effect is that many elderly people have relative hypocalcemia and high serum (PTH) concentrations [8]; this secondary hyperparathyroidism can be attenuated by the administration of physiological doses of vitamin D [9]. However, older persons confined indoors may have low serum calcidiol (25-hydroxyvitamin D) concentrations even if dietary intake is not poor [10]. Vitamin D deficiency appears to be common among other adult populations as well. In a study of 290 patients hospitalized on a general medical service [11], vitamin D deficiency was detected in 164 patients (57 percent), of whom 65 (22 percent) were considered severely deficient (serum concentration of 25-hydroxyvitamin D <8 ng/mL [20 nmol/L]). Inadequate vitamin D intake, winter season, and housebound status were independent predictors of vitamin D deficiency. In a subgroup of 77 patients less than age 65 years without known risk factors, the prevalence of vitamin D deficiency was still 42 percent. Vitamin D deficiency is also common in healthy, young adults. In a study of healthy adults in the Boston area who underwent 25-hydroxyvitamin D testing at the end of winter and summer, 36 percent of 69 subjects ages 18 to 29 were vitamin D deficient, but the prevalence decreased to 4 percent by the end of the summer [12]. Similar seasonal differences were seen in older groups. Nonspecific musculoskeletal pain is a common symptom of osteomalacia, and the prevalence of unrecognized vitamin D deficiency among patients with these symptoms is extremely high. As an example, in a study of 150 subjects with persistent, nonspecific musculoskeletal pain presenting to an inner city health clinic in Minneapolis, 93 percent were vitamin D deficient (serum 25-hydroxyvitamin D concentration < or = 20 ng/mL (50 nmol/L)), and 28 percent of all patients had severe deficiency (concentration < or = 8 ng/mL (20 nmol/L)) [13]. Thus, patients who present with nonspecific musculoskeletal pain should be screened for vitamin D deficiency. Dietary vitamin D deficiency can also occur in children. Among 618 Asian children in the United Kingdom, 27 percent had serum 25-hydroxyvitamin D concentrations <10 ng/mL (25 nmol/L). Serum 25-hydroxyvitamin D concentrations were correlated with ingestion of vitamin D supplements in these children, notwithstanding that increasing skin pigmentation is associated with less cutaneous vitamin D production [14]. (See "Etiology and treatment of hypocalcemic rickets in children"). Patients with advanced cystic fibrosis are usually deficient in vitamin D [15], and they require more than the usual recommended dose for young adults (eg, more than 400 IU/day). Other causes of vitamin D deficiency due to diminished absorption are gastrectomy (which is now performed less often), celiac disease, malabsorption, extensive bowel surgery, inflammatory bowel disease, and advanced cystic fibrosis [15,16]. Although they are uncommon causes of vitamin D deficiency, gastrectomy and celiac sprue have been among the most frequent causes of chronic vitamin D deficiency that becomes clinically evident as osteomalacia [16]. (See "Clinical manifestations and etiology of osteomalacia"). Diminished availability of calcidiol – The vitamin D that reaches the liver is 25-hydroxylated to produce calcidiol (25-hydroxyvitamin D) (show figure 1). This conversion can be impaired in patients with severe liver disease [17] and those taking drugs that increase the activity of P-450 enzymes that inactivate vitamin D, such as anticonvulsants (phenobarbital, phenytoin, carbamazepine), alcohol, isoniazid, theophylline, and rifampin [18-22]. Supplementation with up to 4000 IU of vitamin D per day may be necessary to prevent vitamin D deficiency in these patients [23]. Most of the calcidiol in serum is bound to vitamin D-binding protein. Patients with the nephrotic syndrome can excrete enough vitamin D-binding protein (with calcidiol bound to it) to become vitamin D-deficient, and may develop hypocalcemia and hypophosphatemia [24]. Decreased renal production of calcitriol – The final step in vitamin D metabolism is the 1-hydroxylation of calcidiol in the kidney to produce calcitriol (1,25-dihydroxyvitamin D). This reaction is stimulated by PTH and hypophosphatemia, and inhibited by calcium and phosphate [25]. In patients with renal failure, calcitriol production is low, due mostly to loss of the enzyme but also to hyperphosphatemia; the result is hypocalcemia, hyperparathyroidism, and bone disease. (See "Pathogenesis of renal osteodystrophy"). The term vitamin D-dependent rickets defines two syndromes, both inherited as autosomal recessive traits, characterized by hypocalcemia, hypophosphatemia, and rickets occurring in childhood. Type 1 vitamin D-dependent rickets, also called pseudovitamin D deficiency rickets, is characterized by an inability to produce calcitriol due to an inactivating mutation in the 1-hydroxylase gene. (See "Etiology and treatment of hypocalcemic rickets in children"). Vitamin D resistance – What had been called type 2 vitamin D-dependent rickets is actually a form of vitamin D resistance and is now known as hereditary vitamin D-resistant rickets (HVDRR). It is associated with end-organ resistance to calcitriol due most often to mutations in the gene encoding the vitamin D receptor. (See "Etiology and treatment of hypocalcemic rickets in children").
 on bone, vitamin D has the net effect of increasing the serum calcium and phosphate concentrations (show figure 1). Vitamin D deficiency or resistance interferes with these processes, causing hypocalcemia and hypophosphatemia. The latter is usually more prominent, since hypocalcemia stimulates the release of PTH. The secondary hyperparathyroidism, via its actions on bone and the kidney, partially corrects the hypocalcemia but worsens the hypophosphatemia by increasing urinary phosphate excretion. (See Chapter 6F: Hormonal regulation of calcium and phosphate balance ). This topic will review the major causes of vitamin D deficiency or resistance. The major causes of the clinical manifestations of this problem – hypophosphatemia and hypocalcemia – are discussed separately. (See Causes of hypophosphatemia and see Etiology of hypocalcemia in adults ). ETIOLOGY – Vitamin D deficiency can occur as a result of decreased intake or absorption, reduced sun exposure, increased hepatic catabolism, decreased endogenous synthesis (via 25-hydroxylation in the liver and subsequent 1-hydroxylation in the kidney), or end-organ resistance (show table 1) [1]. (See Metabolism of vitamin D ). Reduced vitamin D intake or production in skin – In the United States, most vitamin D is derived from foods that are rich in the vitamin (fatty fishes) or fortified with the vitamin (milk and related products and cereals). The remainder is synthesized in the skin from 7-dehydrocholesterol under the influence of ultraviolet light (show figure 1). Vitamin D deficiency can therefore occur in people who live without sun exposure (including those whose skin is constantly protected from the sun) and whose dietary intake is low, which is most common in countries distant from the equator in which foods are not fortified with vitamin D [2]. Cutaneous vitamin D production and vitamin D stores decline with age [3]. This change is most prominent in the winter. In temperate areas such as Boston and Edmonton, for example, cutaneous production of vitamin D virtually ceases in winter, especially in the elderly [4,5]. It may also be more common in Asian Indian immigrants to the United States who may have vitamin D deficiency, even with adequate sun exposure [6]. In patients with a history of extensive burn injuries, vitamin D synthesis in skin is below normal, even with sun exposure [7]. Thus, these patients should receive vitamin D supplementation. In addition to reduced endogenous production, vitamin D intake is often low in older subjects. It has been estimated that approximately one-half of elderly women consume less than 137 IU/day of vitamin D, and nearly one-quarter consume less than 65 IU/day (recommended intake 400 IU/day for people 51 to 70 years old and 600 IU/day for people 71 years old and older) [5]. The net effect is that many elderly people have relative hypocalcemia and high serum (PTH) concentrations [8]; this secondary hyperparathyroidism can be attenuated by the administration of physiological doses of vitamin D [9]. However, older persons confined indoors may have low serum calcidiol (25-hydroxyvitamin D) concentrations even if dietary intake is not poor [10]. Vitamin D deficiency appears to be common among other adult populations as well. In a study of 290 patients hospitalized on a general medical service [11], vitamin D deficiency was detected in 164 patients (57 percent), of whom 65 (22 percent) were considered severely deficient (serum concentration of 25-hydroxyvitamin D <8 ng/mL [20 nmol/L]). Inadequate vitamin D intake, winter season, and housebound status were independent predictors of vitamin D deficiency. In a subgroup of 77 patients less than age 65 years without known risk factors, the prevalence of vitamin D deficiency was still 42 percent. Vitamin D deficiency is also common in healthy, young adults. In a study of healthy adults in the Boston area who underwent 25-hydroxyvitamin D testing at the end of winter and summer, 36 percent of 69 subjects ages 18 to 29 were vitamin D deficient, but the prevalence decreased to 4 percent by the end of the summer [12]. Similar seasonal differences were seen in older groups. Nonspecific musculoskeletal pain is a common symptom of osteomalacia, and the prevalence of unrecognized vitamin D deficiency among patients with these symptoms is extremely high. As an example, in a study of 150 subjects with persistent, nonspecific musculoskeletal pain presenting to an inner city health clinic in Minneapolis, 93 percent were vitamin D deficient (serum 25-hydroxyvitamin D concentration < or = 20 ng/mL (50 nmol/L)), and 28 percent of all patients had severe deficiency (concentration < or = 8 ng/mL (20 nmol/L)) [13]. Thus, patients who present with nonspecific musculoskeletal pain should be screened for vitamin D deficiency. Dietary vitamin D deficiency can also occur in children. Among 618 Asian children in the United Kingdom, 27 percent had serum 25-hydroxyvitamin D concentrations <10 ng/mL (25 nmol/L). Serum 25-hydroxyvitamin D concentrations were correlated with ingestion of vitamin D supplements in these children, notwithstanding that increasing skin pigmentation is associated with less cutaneous vitamin D production [14]. (See Etiology and treatment of hypocalcemic rickets in children ). Patients with advanced cystic fibrosis are usually deficient in vitamin D [15], and they require more than the usual recommended dose for young adults (eg, more than 400 IU/day). Other causes of vitamin D deficiency due to diminished absorption are gastrectomy (which is now performed less often), celiac disease, malabsorption, extensive bowel surgery, inflammatory bowel disease, and advanced cystic fibrosis [15,16]. Although they are uncommon causes of vitamin D deficiency, gastrectomy and celiac sprue have been among the most frequent causes of chronic vitamin D deficiency that becomes clinically evident as osteomalacia [16]. (See Clinical manifestations and etiology of osteomalacia ). Diminished availability of calcidiol – The vitamin D that reaches the liver is 25-hydroxylated to produce calcidiol (25-hydroxyvitamin D) (show figure 1). This conversion can be impaired in patients with severe liver disease [17] and those taking drugs that increase the activity of P-450 enzymes that inactivate vitamin D, such as anticonvulsants (phenobarbital, phenytoin, carbamazepine), alcohol, isoniazid, theophylline, and rifampin [18-22]. Supplementation with up to 4000 IU of vitamin D per day may be necessary to prevent vitamin D deficiency in these patients [23]. Most of the calcidiol in serum is bound to vitamin D-binding protein. Patients with the nephrotic syndrome can excrete enough vitamin D-binding protein (with calcidiol bound to it) to become vitamin D-deficient, and may develop hypocalcemia and hypophosphatemia [24]. Decreased renal production of calcitriol – The final step in vitamin D metabolism is the 1-hydroxylation of calcidiol in the kidney to produce calcitriol (1,25-dihydroxyvitamin D). This reaction is stimulated by PTH and hypophosphatemia, and inhibited by calcium and phosphate [25]. In patients with renal failure, calcitriol production is low, due mostly to loss of the enzyme but also to hyperphosphatemia; the result is hypocalcemia, hyperparathyroidism, and bone disease. (See Pathogenesis of renal osteodystrophy ). The term vitamin D-dependent rickets defines two syndromes, both inherited as autosomal recessive traits, characterized by hypocalcemia, hypophosphatemia, and rickets occurring in childhood. Type 1 vitamin D-dependent rickets, also called pseudovitamin D deficiency rickets, is characterized by an inability to produce calcitriol due to an inactivating mutation in the 1-hydroxylase gene. (See Etiology and treatment of hypocalcemic rickets in children ). Vitamin D resistance – What had been called type 2 vitamin D-dependent rickets is actually a form of vitamin D resistance and is now known as hereditary vitamin D-resistant rickets (HVDRR). It is associated with end-organ resistance to calcitriol due most often to mutations in the gene encoding the vitamin D receptor. (See Etiology and treatment of hypocalcemic rickets in children ).")
22
D vitamini eksikliğinin dönemleri
Kalıcı hipokalsemi Plazma fosfat düzeyi belirgin Plazma PTH (Dışarıdan verilen hormon kalsemiyi artırmaz.) Demineralizasyon belirgindir, kemiklerin büyümesi veya yenilenmesi durmuştur. 1. Dönem Hipokalsemi gelişir. PTH salgısı Plazma fosfat düzeyi normal Kemiklerde hafif demineralizasyon Her 3 dönemde de alkalin fosfataz düzeyi yüksektir. Paratiroid bezlerde hiperplazi gelişir. Bu hastalara D vitamini verildiğinde ilk birkaç gün plazma kalsiyum ve fosfat düzeyi daha da düşer; ondan sonra yükselmeye başlayarak normale ulaşır. 2. Dönem Kan kalsiyumu normal Plazma fosfat düzeyi Aminoasidüri Plazma PTH (Dışarıdan verilen hormona kemikler yanıt verir ve hiperkalsemi oluşur.) Belirgin raşitizm veya osteomalasi belirtileri
 Demineralizasyon belirgindir, kemiklerin büyümesi veya yenilenmesi durmuştur. 1. Dönem. Hipokalsemi gelişir. PTH salgısı Plazma fosfat düzeyi normal. Kemiklerde hafif demineralizasyon. Her 3 dönemde de alkalin fosfataz düzeyi yüksektir. Paratiroid bezlerde hiperplazi gelişir. Bu hastalara D vitamini verildiğinde ilk birkaç gün plazma kalsiyum ve fosfat düzeyi daha da düşer; ondan sonra yükselmeye başlayarak normale ulaşır. 2. Dönem. Kan kalsiyumu normal Plazma fosfat düzeyi Aminoasidüri Plazma PTH (Dışarıdan verilen hormona kemikler yanıt verir ve hiperkalsemi oluşur.) Belirgin raşitizm veya osteomalasi belirtileri.")
23
D Vitamini Eksikliği Nedenleri
a) Nutrisyonel D vitamini eksikliği Yetersiz beslenme Yeterince güneş ışığı alamama b) Metabolik D vitamini eksiklikleri Kalıtsal Kalıtsal olmayan
 Nutrisyonel D vitamini eksikliği. Yetersiz beslenme. Yeterince güneş ışığı alamama. b) Metabolik D vitamini eksiklikleri. Kalıtsal. Kalıtsal olmayan.")
24
Metabolik D Vitamini Eksikliği
Kalıtsal a. X kromozomuna bağlı dominant D vitaminine dirençli raşitizm (hipofosfatemik raşitizm) Yüksek dozda D vitamini ( Ü/gün) ve oral fosfat (1-3 g/gün) ile tedavi edilir. b. Otozomal resesif D vitaminine bağımlı tip I raşitizm D vitamininin biyoaktivasyonundaki defekte bağlı olarak kalsitriol sentezi bozulmuştur. Fizyolojik dozlardaki kalsitriol (1-2 g/gün) ile tedavi edilir. c. Otozomal resesif tip II raşitizm Kalsitriol reseptörlerinde anormallik vardır. Reseptörün kalsitriole bağlanmasında ya da bağlandıktan sonra kompleksin DNA’ya bağlanmasında bir bozukluk söz konusudur. Yüksek dozdaki D vitaminine veya kalsitriole yanıt vermez. Parenteral kalsiyum tedavisi gerekir. Kalıtsal olmayan Renal osteodistrofi Dışarıdan yeterli miktarda D vitamini verilse de böbrekte ,25 (OH)2D3'e dönüşemez. Kalsitriol ile tedavi edilir. X-LINKED – X-linked hypophosphatemic rickets (XLH) is a dominant disorder with a prevalence of approximately one case per 20,000 individuals [1,2]. Pathogenesis – The pathogenesis of X-linked hypophosphatemic rickets is not fully understood. A number of functional studies indicate that the tubular defect in patients with XLH and in the corresponding mouse model (Hyp mouse) is due to a circulating factor rather than to a defect in the kidney [3,4]. These studies also suggest the existence of a primary osteoblast abnormality in addition to the renal tubular defect: • Cross-transplantation studies showed that the Hyp mouse phenotype was neither corrected nor transferred by renal transplantation [3]. • When the circulation of Hyp mice and normal mice were surgically joined, the normal mice developed enhanced renal phosphate excretion and hypophosphatemia, indicating the importance of a circulating factor [4]. • Osteoblasts from Hyp mice that were transplanted into normal mice produced abnormal bone suggesting a primary osteoblast abnormality [5-7]. • Compared with immortalized osteoblasts from SV40 transgenic normal mice, osteoblast cultures from SV40 hyp-mice exhibit diminished calcium accumulation into extracellular matrix and reduced mineralization nodules [7]. The gene responsible for XLH was identified on chromosome Xp22.1 and was named PHEX (Phosphate regulating Endopeptidase on the X chromosome) [8]. This gene codes for a cell surface-bound protein-cleaving enzyme. PHEX is expressed predominantly in bone and teeth [9], but is also found in organs that apparently do not express any phenotypic abnormalities in patients with XLH (such as lung, brain, muscle, ovary, and testis) [10,11]. A large number of mutations in the PHEX gene can cause XLH and there is no obvious correlation between genotype and phenotype [12,13]. A list of PHEX mutations can be found at Mutations in the PHEX gene in the tissues where it is expressed lead to a posttranscriptional defect in the kidney that results in underexpression of the sodium phosphate cotransporter that is responsible for phosphate reabsorption in the proximal tubule [14,15]. The current view is that, under normal conditions, the product of the PHEX gene degrades and inactivates hormone-like substances (the "circulating factor" described above, also called phosphatonins) that promote phosphate excretion and impair bone mineralization [7]. In patients with XLH, inactivating mutations of PHEX probably result in a failure to inactivate the phosphatonins, leading to abnormally high circulating concentrations of these factors and consequent excessive phosphate excretion and abnormal mineralization. A number of hormones that may serve as phosphatonins have been identified. Among these are fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPE) [16], and frizzled related protein 4 (FRP-4) [17,18], all of which exhibit some (but not all) of the biological properties of the putative phosphatonin(s) [19]. The hypothesis that FGF23 is a circulating phosphatonin is supported by the role that FGF23 plays in other forms of hypophophosphatemic rickets. In the autosomal dominant form, mutations in the FGF23 gene interfere with its cleavage [20,21], whereas overproduction of FGF23 in tumors can be responsible for tumor-induced osteomalacia [22]. Thus, it has been suggested that elevated levels of FGF23 may be the final common pathway for these three phosphate-wasting disorders [23]. However, it has not yet been shown whether PHEX cleaves FGF23. Furthermore, observations in Hyp mice suggest that the circulating substance(s) are produced by osteoblasts [5-7,24,25], and osteoblasts do not make FGF23 [26]. These findings suggest that other sites of PHEX expression or other phosphaturic factors may be important [26]. The possibility that MEPE and FRP-4 might serve as phosphatonin(s) derived from the discovery of overproduction of these factors by tumors in patients with tumor-induced osteomalacia [16,17]. Moreover, the biological properties of these factors, both in vitro and in vivo, are remarkably similar to those ascribed to phosphatonin. To date, however, none of these factors has been unequivocally identified as phosphatonin and further study will be necessary to better define this issue. Treatment – The goals of therapy for XLH depend upon the skeletal maturity of the patient. In skeletally immature patients, the goals of therapy are normal longitudinal growth rate and the achievement of normal serum alkaline phosphatase concentrations, rather than normalization of serum phosphate concentration. On the other hand, in skeletally mature patients, the goal of therapy is to prevent symptoms of hypophosphatemia, such as muscle weakness and paresthesias. Children – The current therapy for XLH has been used for more than 20 years and consists of the oral administration of phosphate and calcitriol [2,31]. The administration of phosphate increases the plasma phosphate concentration, which lowers the plasma ionized calcium concentration, and further reduces the plasma calcitriol concentration (by removing the hypophosphatemic stimulus to its synthesis). This causes secondary hyperparathyroidism due both to hypocalcemia and removal of the normal inhibitory effect of calcitriol on parathyroid hormone (PTH) synthesis. The elevated PTH levels can aggravate the bone disease and increase urinary phosphate excretion, thereby defeating the aim of oral therapy. The administration of calcitriol is necessary to increase the intestinal absorption of calcium, and to a lesser degree phosphate in an effort to prevent secondary hyperparathyroidism. • Calcitriol is administered in two doses per day (10 to 20 ng/kg per dose) • Phosphorus is administered in four to five doses that are equally spaced over the 24-hour period; the starting dose is 40 mg of elemental phosphorus/kg per day. In our experience, a nighttime dose is important to achieve satisfactory results. Some catch-up growth should be noticeable within the first year of therapy. If this does not occur despite good compliance, the daily phosphorus dose should be increased in steps of 250 mg to 500 mg up to a maximum of 3500 mg/day. Various forms of phosphate salts are available (eg, sodium phosphate, potassium phosphate) with no obvious advantage of one preparation over another. Tablets usually contain 250 mg elemental phosphorus per pill. Children who cannot take pills can receive phosphorus supplementation in the form of Joulie's solution (155 g of dibasic anhydrous sodium phosphate and 64 g of phosphoric acid 85 percent per liter solution, corresponding to 50 mg/mL of elemental phosphorus). The amount of phosphorus supplementation is usually limited by the occurrence of diarrhea. If diarrhea is a problem, the dosage of phosphorus should be decreased by 250 to 500 mg and then gradually re-increased in steps of 125 mg. The aim should be to administer the minimum amount of phosphorus that is sufficient for normal growth. Slow growth and persistently elevated alkaline phosphatase activity indicate inadequate dose of phosphorus or compliance with therapy. Children should be seen every three months to monitor height, serum concentrations of calcium, phosphate, alkaline phosphatase, creatinine, and urinary calcium excretion. Renal ultrasonography should be performed once per year to evaluate nephrocalcinosis (see below). A hand radiograph should be obtained once per year to exclude the reappearance of rickets and to determine bone age. In the majority of prepubertal children who are treated with this regimen, radiological signs of rickets disappear, growth improves [32,33], and deformities of the lower limbs are prevented or corrected [31]. Other effects of therapy include elevation in the plasma phosphate concentration, increase in urinary calcium excretion, marked improvement in bone or joint pain, and a significant decrease in osteoid thickness and mean osteoid volume [34]. The characteristic hypomineralized periosteocytic lesions persist on biopsy even though the histologic appearance of osteomalacia improves [30]. This observation supports the hypothesis that a primary osteoblast defect contributes to the pathogenesis of XLH [5]. Since the aim of treatment is to achieve normal growth, therapy is maintained as long as the growth plates are open (usually up to the age of 15 to 17 years). Adults – It is controversial whether adults should continue therapy on a routine basis. Histologic evidence of osteomalacia recurs after phosphorus and calcitriol are stopped [30]. However, most of these patients have few if any skeletal complaints. Such patients may not benefit from therapy, whereas they may experience complications (see below). As a result, it may be preferable to restrict treatment to those patients who are symptomatic. • Phosphate is administered in a dose of 1 to 4 g/day in three to four divided doses In contrast to children, once a patient reaches adult height and the epiphyses have fused, the goal of therapy is simply to prevent symptoms of hypophosphatemia, such as muscle weakness and paresthesias. The minimal dose to achieve this goal should be sought in each patient, and many patients will require no therapy at all. Adults should be monitored at least annually for serum phosphorus, calcium, creatinine, and PTH. Less clear is whether therapy should be resumed in affected women who become pregnant. However, since the plasma phosphate of the fetus is determined by diffusion across the placenta, it seems reasonable to maintain a higher level of phosphate than that in the untreated state during pregnancy. Studies are currently underway to determine if hypophosphatemia during pregnancy affects bone mineralization or development in the unborn. Complications – There are two important complications of the treatment of XLH: nephrocalcinosis and hyperparathyroidism. • Nephrocalcinosis – Nephrocalcinosis can be demonstrated on renal ultrasonography in up to 80 percent of patients with XLH and is associated with renal tubular acidosis [34,35]. The renal calcifications are located primarily in the tubules and are composed exclusively of calcium phosphate [36]. The degree of calcium phosphate deposition correlates with the mean phosphate dose but not with the dose of calcitriol or the duration of therapy [34,36]. Although most patients have a normal plasma creatinine concentration, the long-term effect of nephrocalcinosis on renal function is not known. Isolated cases of renal insufficiency have been reported [36]. The development of nephrocalcinosis is probably linked to intermittent episodes of hypercalcemia and hypercalciuria. These can result from an excessive calcitriol dosage or from noncompliance with oral phosphate supplementation [37]. Thus, careful monitoring and control of serum and urine calcium is necessary to minimize nephrocalcinosis. The dose of calcitriol should be reduced when hypercalcemia or hypercalciuria are present. The importance of strict adherence to the burdensome phosphorus supplementation schedule must be repeatedly emphasized to the patients and their caretakers. Studies in Hyp mice suggest that nonhypercalcemic analogues of calcitriol, such as 22-oxacalcitriol, may provide a similar increase in plasma phosphate without producing hypercalcemia or hypercalciuria [7]. These analogues have not been evaluated in humans. • Hyperparathyroidism – Hyperparathyroidism has been reported in XLH. This usually occurs after years of treatment, but also may be present early in the course of the disease [38]. It is thought that complexing of calcium with phosphate results in intermittent hypocalcemia and persistent stimulation of parathyroid hormone (PTH) release despite the administration of calcitriol. When this secondary hyperparathyroidism is not adequately controlled, autonomous (tertiary) hyperparathyroidism can occur, necessitating surgical intervention [39,40]. Adjuvant therapy – Several forms of adjuvant therapy are being tested to improve the efficacy or diminish side effects of the current therapy of XLH. The administration of recombinant human growth hormone can improve short-term growth in children with XLH [41]. This may translate into taller final height, as shown in a long-term study on six patients [42]. However, it is possible that treatment with growth hormone aggravates the preexistent disproportionate stature of such children [43]. Currently available data do not support the recommendation of growth hormone therapy outside of the study setting. An observational study of 11 children with XLH showed that the addition of hydrochlorothiazide decreased urinary calcium excretion and prevented progression of nephrocalcinosis during the 3.3-year observation period [44]. This approach may be useful in the management of hypophosphatemic rickets if the results are confirmed in other studies. A placebo-controlled trial on 15 patients with XLH tested the value of 24,25-dihydroxyvitamin D as a supplement to current standard treatment [45]. The main effect was improved control of hyperparathyroidism. Unfortunately, 24,25 dihydroxyvitamin D is currently not available as a pharmaceutical agent. (See "Metabolism of vitamin D").
 Yüksek dozda D vitamini ( Ü/gün) ve oral fosfat (1-3 g/gün) ile tedavi edilir. b. Otozomal resesif D vitaminine bağımlı tip I raşitizm. D vitamininin biyoaktivasyonundaki defekte bağlı olarak kalsitriol sentezi bozulmuştur. Fizyolojik dozlardaki kalsitriol (1-2 g/gün) ile tedavi edilir. c. Otozomal resesif tip II raşitizm Kalsitriol reseptörlerinde anormallik vardır. Reseptörün kalsitriole bağlanmasında ya da bağlandıktan sonra kompleksin DNA’ya bağlanmasında bir bozukluk söz konusudur. Yüksek dozdaki D vitaminine veya kalsitriole yanıt vermez. Parenteral kalsiyum tedavisi gerekir. Kalıtsal olmayan. Renal osteodistrofi Dışarıdan yeterli miktarda D vitamini verilse de böbrekte 1,25 (OH)2D3 e dönüşemez. Kalsitriol ile tedavi edilir. X-LINKED – X-linked hypophosphatemic rickets (XLH) is a dominant disorder with a prevalence of approximately one case per 20,000 individuals [1,2]. Pathogenesis – The pathogenesis of X-linked hypophosphatemic rickets is not fully understood. A number of functional studies indicate that the tubular defect in patients with XLH and in the corresponding mouse model (Hyp mouse) is due to a circulating factor rather than to a defect in the kidney [3,4]. These studies also suggest the existence of a primary osteoblast abnormality in addition to the renal tubular defect: • Cross-transplantation studies showed that the Hyp mouse phenotype was neither corrected nor transferred by renal transplantation [3]. • When the circulation of Hyp mice and normal mice were surgically joined, the normal mice developed enhanced renal phosphate excretion and hypophosphatemia, indicating the importance of a circulating factor [4]. • Osteoblasts from Hyp mice that were transplanted into normal mice produced abnormal bone suggesting a primary osteoblast abnormality [5-7]. • Compared with immortalized osteoblasts from SV40 transgenic normal mice, osteoblast cultures from SV40 hyp-mice exhibit diminished calcium accumulation into extracellular matrix and reduced mineralization nodules [7]. The gene responsible for XLH was identified on chromosome Xp22.1 and was named PHEX (Phosphate regulating Endopeptidase on the X chromosome) [8]. This gene codes for a cell surface-bound protein-cleaving enzyme. PHEX is expressed predominantly in bone and teeth [9], but is also found in organs that apparently do not express any phenotypic abnormalities in patients with XLH (such as lung, brain, muscle, ovary, and testis) [10,11]. A large number of mutations in the PHEX gene can cause XLH and there is no obvious correlation between genotype and phenotype [12,13]. A list of PHEX mutations can be found at Mutations in the PHEX gene in the tissues where it is expressed lead to a posttranscriptional defect in the kidney that results in underexpression of the sodium phosphate cotransporter that is responsible for phosphate reabsorption in the proximal tubule [14,15]. The current view is that, under normal conditions, the product of the PHEX gene degrades and inactivates hormone-like substances (the circulating factor described above, also called phosphatonins) that promote phosphate excretion and impair bone mineralization [7]. In patients with XLH, inactivating mutations of PHEX probably result in a failure to inactivate the phosphatonins, leading to abnormally high circulating concentrations of these factors and consequent excessive phosphate excretion and abnormal mineralization. A number of hormones that may serve as phosphatonins have been identified. Among these are fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPE) [16], and frizzled related protein 4 (FRP-4) [17,18], all of which exhibit some (but not all) of the biological properties of the putative phosphatonin(s) [19]. The hypothesis that FGF23 is a circulating phosphatonin is supported by the role that FGF23 plays in other forms of hypophophosphatemic rickets. In the autosomal dominant form, mutations in the FGF23 gene interfere with its cleavage [20,21], whereas overproduction of FGF23 in tumors can be responsible for tumor-induced osteomalacia [22]. Thus, it has been suggested that elevated levels of FGF23 may be the final common pathway for these three phosphate-wasting disorders [23]. However, it has not yet been shown whether PHEX cleaves FGF23. Furthermore, observations in Hyp mice suggest that the circulating substance(s) are produced by osteoblasts [5-7,24,25], and osteoblasts do not make FGF23 [26]. These findings suggest that other sites of PHEX expression or other phosphaturic factors may be important [26]. The possibility that MEPE and FRP-4 might serve as phosphatonin(s) derived from the discovery of overproduction of these factors by tumors in patients with tumor-induced osteomalacia [16,17]. Moreover, the biological properties of these factors, both in vitro and in vivo, are remarkably similar to those ascribed to phosphatonin. To date, however, none of these factors has been unequivocally identified as phosphatonin and further study will be necessary to better define this issue. Treatment – The goals of therapy for XLH depend upon the skeletal maturity of the patient. In skeletally immature patients, the goals of therapy are normal longitudinal growth rate and the achievement of normal serum alkaline phosphatase concentrations, rather than normalization of serum phosphate concentration. On the other hand, in skeletally mature patients, the goal of therapy is to prevent symptoms of hypophosphatemia, such as muscle weakness and paresthesias. Children – The current therapy for XLH has been used for more than 20 years and consists of the oral administration of phosphate and calcitriol [2,31]. The administration of phosphate increases the plasma phosphate concentration, which lowers the plasma ionized calcium concentration, and further reduces the plasma calcitriol concentration (by removing the hypophosphatemic stimulus to its synthesis). This causes secondary hyperparathyroidism due both to hypocalcemia and removal of the normal inhibitory effect of calcitriol on parathyroid hormone (PTH) synthesis. The elevated PTH levels can aggravate the bone disease and increase urinary phosphate excretion, thereby defeating the aim of oral therapy. The administration of calcitriol is necessary to increase the intestinal absorption of calcium, and to a lesser degree phosphate in an effort to prevent secondary hyperparathyroidism. • Calcitriol is administered in two doses per day (10 to 20 ng/kg per dose) • Phosphorus is administered in four to five doses that are equally spaced over the 24-hour period; the starting dose is 40 mg of elemental phosphorus/kg per day. In our experience, a nighttime dose is important to achieve satisfactory results. Some catch-up growth should be noticeable within the first year of therapy. If this does not occur despite good compliance, the daily phosphorus dose should be increased in steps of 250 mg to 500 mg up to a maximum of 3500 mg/day. Various forms of phosphate salts are available (eg, sodium phosphate, potassium phosphate) with no obvious advantage of one preparation over another. Tablets usually contain 250 mg elemental phosphorus per pill. Children who cannot take pills can receive phosphorus supplementation in the form of Joulie s solution (155 g of dibasic anhydrous sodium phosphate and 64 g of phosphoric acid 85 percent per liter solution, corresponding to 50 mg/mL of elemental phosphorus). The amount of phosphorus supplementation is usually limited by the occurrence of diarrhea. If diarrhea is a problem, the dosage of phosphorus should be decreased by 250 to 500 mg and then gradually re-increased in steps of 125 mg. The aim should be to administer the minimum amount of phosphorus that is sufficient for normal growth. Slow growth and persistently elevated alkaline phosphatase activity indicate inadequate dose of phosphorus or compliance with therapy. Children should be seen every three months to monitor height, serum concentrations of calcium, phosphate, alkaline phosphatase, creatinine, and urinary calcium excretion. Renal ultrasonography should be performed once per year to evaluate nephrocalcinosis (see below). A hand radiograph should be obtained once per year to exclude the reappearance of rickets and to determine bone age. In the majority of prepubertal children who are treated with this regimen, radiological signs of rickets disappear, growth improves [32,33], and deformities of the lower limbs are prevented or corrected [31]. Other effects of therapy include elevation in the plasma phosphate concentration, increase in urinary calcium excretion, marked improvement in bone or joint pain, and a significant decrease in osteoid thickness and mean osteoid volume [34]. The characteristic hypomineralized periosteocytic lesions persist on biopsy even though the histologic appearance of osteomalacia improves [30]. This observation supports the hypothesis that a primary osteoblast defect contributes to the pathogenesis of XLH [5]. Since the aim of treatment is to achieve normal growth, therapy is maintained as long as the growth plates are open (usually up to the age of 15 to 17 years). Adults – It is controversial whether adults should continue therapy on a routine basis. Histologic evidence of osteomalacia recurs after phosphorus and calcitriol are stopped [30]. However, most of these patients have few if any skeletal complaints. Such patients may not benefit from therapy, whereas they may experience complications (see below). As a result, it may be preferable to restrict treatment to those patients who are symptomatic. • Phosphate is administered in a dose of 1 to 4 g/day in three to four divided doses. In contrast to children, once a patient reaches adult height and the epiphyses have fused, the goal of therapy is simply to prevent symptoms of hypophosphatemia, such as muscle weakness and paresthesias. The minimal dose to achieve this goal should be sought in each patient, and many patients will require no therapy at all. Adults should be monitored at least annually for serum phosphorus, calcium, creatinine, and PTH. Less clear is whether therapy should be resumed in affected women who become pregnant. However, since the plasma phosphate of the fetus is determined by diffusion across the placenta, it seems reasonable to maintain a higher level of phosphate than that in the untreated state during pregnancy. Studies are currently underway to determine if hypophosphatemia during pregnancy affects bone mineralization or development in the unborn. Complications – There are two important complications of the treatment of XLH: nephrocalcinosis and hyperparathyroidism. • Nephrocalcinosis – Nephrocalcinosis can be demonstrated on renal ultrasonography in up to 80 percent of patients with XLH and is associated with renal tubular acidosis [34,35]. The renal calcifications are located primarily in the tubules and are composed exclusively of calcium phosphate [36]. The degree of calcium phosphate deposition correlates with the mean phosphate dose but not with the dose of calcitriol or the duration of therapy [34,36]. Although most patients have a normal plasma creatinine concentration, the long-term effect of nephrocalcinosis on renal function is not known. Isolated cases of renal insufficiency have been reported [36]. The development of nephrocalcinosis is probably linked to intermittent episodes of hypercalcemia and hypercalciuria. These can result from an excessive calcitriol dosage or from noncompliance with oral phosphate supplementation [37]. Thus, careful monitoring and control of serum and urine calcium is necessary to minimize nephrocalcinosis. The dose of calcitriol should be reduced when hypercalcemia or hypercalciuria are present. The importance of strict adherence to the burdensome phosphorus supplementation schedule must be repeatedly emphasized to the patients and their caretakers. Studies in Hyp mice suggest that nonhypercalcemic analogues of calcitriol, such as 22-oxacalcitriol, may provide a similar increase in plasma phosphate without producing hypercalcemia or hypercalciuria [7]. These analogues have not been evaluated in humans. • Hyperparathyroidism – Hyperparathyroidism has been reported in XLH. This usually occurs after years of treatment, but also may be present early in the course of the disease [38]. It is thought that complexing of calcium with phosphate results in intermittent hypocalcemia and persistent stimulation of parathyroid hormone (PTH) release despite the administration of calcitriol. When this secondary hyperparathyroidism is not adequately controlled, autonomous (tertiary) hyperparathyroidism can occur, necessitating surgical intervention [39,40]. Adjuvant therapy – Several forms of adjuvant therapy are being tested to improve the efficacy or diminish side effects of the current therapy of XLH. The administration of recombinant human growth hormone can improve short-term growth in children with XLH [41]. This may translate into taller final height, as shown in a long-term study on six patients [42]. However, it is possible that treatment with growth hormone aggravates the preexistent disproportionate stature of such children [43]. Currently available data do not support the recommendation of growth hormone therapy outside of the study setting. An observational study of 11 children with XLH showed that the addition of hydrochlorothiazide decreased urinary calcium excretion and prevented progression of nephrocalcinosis during the 3.3-year observation period [44]. This approach may be useful in the management of hypophosphatemic rickets if the results are confirmed in other studies. A placebo-controlled trial on 15 patients with XLH tested the value of 24,25-dihydroxyvitamin D as a supplement to current standard treatment [45]. The main effect was improved control of hyperparathyroidism. Unfortunately, 24,25 dihydroxyvitamin D is currently not available as a pharmaceutical agent. (See Metabolism of vitamin D ).")
25
Vitamin D Preparatları
Kolekalsiferol (D3 vit) DEVİT İÜ ampul 1,72 YTL 3333 İÜ/1 mL (25 damla), 15 mL/şişe 2,05 YTL D3 vitamini + Kalsiyum CALCİMAX D3 880 İÜ + 1 g, 40 efervesan tablet 27,56 YTL CAL-D-VİTA 400 İÜ mg, 10 efervesan tablet 4,13YTL 400 İÜ mg, 30 efervesan tablet 10,42 YTL FOSFOKALSİYUM 400 İÜ + 0,6 g kalsiyum glukonat + 0,2 g kalsiyum laktat + 0,2 g kalsiyum fosfat /ölçek, 75 g granül 2,31 YTL Ergokalsiferol (D2 vit) CALCİDİNE 125 mg kalsiyum magnezyum inozitohekzofosfat mg kalsiyum glukonat IU Vitamin D2/5 g, 75 g kutu 2,43 YTL
 DEVİT İÜ ampul 1,72 YTL 3333 İÜ/1 mL (25 damla), 15 mL/şişe 2,05 YTL. D3 vitamini + Kalsiyum. CALCİMAX D3 880 İÜ + 1 g, 40 efervesan tablet 27,56 YTL. CAL-D-VİTA 400 İÜ mg, 10 efervesan tablet 4,13YTL 400 İÜ mg, 30 efervesan tablet 10,42 YTL. FOSFOKALSİYUM 400 İÜ + 0,6 g kalsiyum glukonat + 0,2 g kalsiyum laktat + 0,2 g kalsiyum fosfat /ölçek, 75 g granül 2,31 YTL. Ergokalsiferol (D2 vit) CALCİDİNE 125 mg kalsiyum magnezyum inozitohekzofosfat mg kalsiyum glukonat IU Vitamin D2/5 g, 75 g kutu 2,43 YTL.")
26
Vitamin D Preparatları
Alfakalsidol (1-hidroksikolekalsiferol) ALPHA D g, 50 kapsül 16,36 YTL g, 100 kapsül 32,75 YTL 1 g, 50 kapsül 37,99 YTL ONE-ALPHA 0.25 g, 30 kapsül 9,39 YTL g, 50 kapsül 15,61 YTL g, 100 kapsül 31,03 YTL 1 g, 30 kapsül 27,80 YTL 1 g, 100 kapsül 92,91 YTL 1 g, 10 ampül 75,13 YTL 2 g, 10 ampül 145,50 YTL Kalsitriol (1,25-dihidroksikolekalsiferol) CALCİJEX 1 g, 25 ampül 180,26 YTL 2 g, 25 ampül 299,74 YTL DECOSTRİOL 0.25 g, 30 kapsül Ruhsatı İptal 0.25 g, 100 kapsül Ruhsatı İptal g, 30 kapsül Ruhsatı İptal g, 100 kapsül Ruhsatı İptal ROCALTROL 0.25 g, 30 kapsül 21,27 YTL g, 100 kapsül 66,99 YTL 0.50 g, 30 kapsül 34,67 YTL 0.50 g, 100 kapsül 109,37 YTL OSTEOD 0.25 g, 30 kapsül 17,43 YTL g, 100 kapsül 28,61 YTL 0.50 g, 30 kapsül 55,13 YTL 0.50 g, 100 kapsül 90,31 YTL Perikalsitol ZEMPLAR 5 micg/1 mL, 5 ampul 217,42 YTL 10 micg/2 mL, 5 ampul 411,17 YTL Sentetik bir D vitamini analoğudur. Kronik böbrek yetersizliği ile ilişkili ikincil hiperparatiroidizmin önlenmesi ve tedavisi için onaylanmıştır.
 ALPHA D g, 50 kapsül 16,36 YTL 0.25 g, 100 kapsül 32,75 YTL 1 g, 50 kapsül 37,99 YTL. ONE-ALPHA 0.25 g, 30 kapsül 9,39 YTL 0.25 g, 50 kapsül 15,61 YTL 0.25 g, 100 kapsül 31,03 YTL 1 g, 30 kapsül 27,80 YTL 1 g, 100 kapsül 92,91 YTL 1 g, 10 ampül 75,13 YTL 2 g, 10 ampül 145,50 YTL. Kalsitriol (1,25-dihidroksikolekalsiferol) CALCİJEX 1 g, 25 ampül 180,26 YTL 2 g, 25 ampül 299,74 YTL. DECOSTRİOL 0.25 g, 30 kapsül Ruhsatı İptal 0.25 g, 100 kapsül Ruhsatı İptal 0.50 g, 30 kapsül Ruhsatı İptal 0.50 g, 100 kapsül Ruhsatı İptal. ROCALTROL 0.25 g, 30 kapsül 21,27 YTL 0.25 g, 100 kapsül 66,99 YTL 0.50 g, 30 kapsül 34,67 YTL 0.50 g, 100 kapsül 109,37 YTL. OSTEOD 0.25 g, 30 kapsül 17,43 YTL 0.25 g, 100 kapsül 28,61 YTL 0.50 g, 30 kapsül 55,13 YTL 0.50 g, 100 kapsül 90,31 YTL. Perikalsitol. ZEMPLAR 5 micg/1 mL, 5 ampul 217,42 YTL 10 micg/2 mL, 5 ampul 411,17 YTL. Sentetik bir D vitamini analoğudur. Kronik böbrek yetersizliği ile ilişkili ikincil hiperparatiroidizmin önlenmesi ve tedavisi için onaylanmıştır.")
27
D Hipervitaminozu (D vitamini zehirlenmesi)
Yüksek dozda D vitamini verilmesi ile ortaya çıkan hiperkalsemi halidir. Ortaya çıkışı verilme süresi ve doza bağlıdır. Bebeklerde hiperkalseminin uzun sürmesi mental ve fiziksel gelişmede gerileme yapar. Gebelerde gelişen D hipervitaminozu fötusta şekil bozukluklarına ve fötal hipoparatiroidizm oluşması sonucu yenidoğanda hipokalsemi, tetani ve konvülsiyonlara neden olur.

28
Kalsitonin Tiroid bezinin parafolliküler hücreleri (C hücreleri) tarafından salgılanır. 32 amino asitli polipeptiddir. Prekürsör moleküldeki 21 aminoasitlik bir bölüm de kalsitonin ile birlikte salgılanır; katakalsin adı verilen bu peptid kalsitonin gibi kalsemiyi düşürür. Parathormonun fizyolojik antagonistidir. Akut verilişi hipokalsemi ve hipofosfatemiye neden olur. Hiperkalsemi (hiperparatiroidizm, hiperkalsemi yapan tümörler vb.'ne bağlı) tedavisinde kullanılır. Son zamanlarda somon kalsitoninin (salkatonin) intranazal uygulanan nazal sprey şekli çıkartılmıştır. İ.M., S.C. ve nazal sprey şeklinde kullanılırlar.
 tedavisinde kullanılır. Son zamanlarda somon kalsitoninin (salkatonin) intranazal uygulanan nazal sprey şekli çıkartılmıştır. İ.M., S.C. ve nazal sprey şeklinde kullanılırlar.")
29
Kalsitonin salgılanması kan kalsiyum düzeyi ve gastrin tarafından düzenlenir
Kalsiyum : Kalsitonin salgılanması Kalsiyum : Kalsitonin salgılanması Gastrin : Kalsitonin salgılanması

30
Kalsitonin Preparatları
Somon Kalsitonin/Salkatonin CALCİTONİNA HUBBER 50 İÜ, 14 doz nazal sprey 36,86 YTL 200 İÜ, 14 doz nazal sprey 66,22 YTL 100 İÜ, 7 doz nazal sprey 48,53 YTL 100 İÜ, 10 ampul 48,53 YTL CALCYNAR 100 İÜ, 7 doz nazal sprey 28,93 YTL 200 İÜ, 7 doz nazal sprey 55,65 YTL MİACALCİC 200 İÜ, 14 doz nazal sprey 66,22 YTL 200 İÜ, 28 doz nazal sprey 119,28 YTL 100 İÜ, 5 ampul 53,19 YTL SALMOCALCİN 100 İÜ, 28 doz nazal sprey 61,1 YTL 100 İÜ, 5 ampul 20,50 YTL TONOCALCİN 100 İÜ, 28 doz nazal sprey 53,01 YTL 200 İÜ, 14 doz nazal sprey 57,31 YTL 200 İÜ, 28 doz nazal sprey 95,72 YTL 50 İÜ, 5 ampul 10,58 YTL 100 İÜ, 5 ampul 19,91 YTL UCECAL 100 İÜ, 5 ampul Ruhsatı iptal

31
Kalsitoninin Endikasyonları
Hiperparatiroidizm ve tümörle ilişkili osteolitik kemik hastalığı gibi durumlarda gelişen hiperkalsemiler Postmenapozal osteoporoz (kronik tedavi için pahalı bir yaklaşım) Paget hastalığı (osteitis deformans)
 Paget hastalığı (osteitis deformans)")
32
Bisfosfonatlar Kemik kristalleri içinde toplanırlar. Pirofosfatlara benzerler. Pirofosfatlarda P-O-P bağı bulunurken, bisfosfonatlarda P-C-P bağı bulunur; bu nedenle enzimatik hidrolize dayanıklıdırlar. İskeletteki yarılanma ömürleri uzundur. Osteoklastların bisfosfonat içeren kristalleri fagosite etmesi, onların metabolik etkinliğini inhibe eder ve böylece kemik rezorbe etme yetenekleri azalır. Antirezorptif etki güçleri kalsitoninden zayıftır. Bisfosfonat Pirofosfat

33
Bisfosfonatlar İlaç olarak ilk kullanılan disodyum etidronattır.
Osteoporoz tedavisindeki dozu 400 mg/gün x 14 gün/3 ayda bir Mineralizasyonu önlediği ve osteomalasi eğilimi yarattığından, bu sakıncasını en aza indirmek için sürekli değil, periyodik verilir. Nefrotoksiktir. Sadece rezorpsiyonu inhibe eden, fakat çökmeye dokunmayan yeni bisfosfonat türevleri: Alendronat, Pamidronat, Klodronat, Risedronat, Tiludronat, İbandronat

34
ACTONEL 5 mg, 28 tablet 49,82 YTL 35 mg, 4 tablet 56,54 YTL Alendronat
Risedronat ACTONEL 5 mg, 28 tablet 49,82 YTL 35 mg, 4 tablet 56,54 YTL Alendronat ANDANTE 10 mg, 28 tablet 49,20 YTL 70 mg, 4 tablet 39,40 YTL BONACTON 70 mg, 4 tablet 36,21 YTL BONEMAX 10 mg, 28 tablet 36,84 YTL 70 mg, 4 tablet 36,84 YTL FOSAMAX 10 mg, 28 tablet 39,46 YTL 70 mg, 4 tablet 39,46 YTL FOSAVANCE 70 mg, 4 tablet 63,86 YTL IU vit D OSALEN 10 mg, 28 tablet 49,15 YTL 70 mg, 4 tablet 36,41 YTL OSTEOMAX 10 mg, 28 tablet 36,71 YTL 70 mg, 4 tablet 36,71 YTL VEGABON 70 mg, 4 tablet 35,01 YTL Etidronat DİDRONAT 200 mg, 60 tablet 26,84 YTL 400 mg, 30 tablet 29,86 YTL Disodyum Klodronat BONEFOS 300 mg/5 mL, 5 ampul 99,60 YTL 400 mg, 100 kapsül 252,80 YTL 800 mg, 60 kapsül 395,84 YTL Gastrik iritasyona neden olması nedeniyle pamidronatın oral formu yoktur. Etidronat hariç tüm bisfosfonatlarda bu yan etki görülmektedir. Pamidronat AREDIA 90 mg/10 mL, 1 flakon 468,08 YTL Zoledronat ZOLENAT 4 mg/5 mL IV inf. sol. 488,10YTL ZOMETA IV 4 mg flakon 602,18 YTL

35
Farmakokinetik Oral alınan dozun %10’undan azı absorbe edilir
Emilime uğrayan bisfosfonatların yarısı kemikte tutulur, kalanı böbrekler yoluyla değişikliğe uğramadan atılır. 1: Ann Med Feb;29(1): Links Bisphosphonates: preclinical aspects and use in osteoporosis. Fleisch HA. Department of Pathophysiology, University of Bern, Switzerland. The bisphosphonates are synthetic compounds characterized by a P-C-P bond. They have a strong affinity to calcium phosphates and hence to bone mineral. In vitro they inhibit both formation and dissolution of the latter. Many of the bisphosphonates inhibit bone resorption, the newest compounds being 10,000 times more active than etidronate, the first bisphosphonate described. The antiresorbing effect is cell mediated, partly by a direct action on the osteoclasts, partly through the osteoblasts, which produce an inhibitor of osteoclastic recruitment. When given in large amounts, some bisphosphonates can also inhibit normal and ectopic mineralization through a physical-chemical inhibition of crystal growth. In the growing rat the inhibition of resorption is accompanied by an increase in intestinal absorption and an increased balance of calcium. Bisphosphonates also prevent various types of experimental osteoporosis, such as after immobilization, ovariectomy, orchidectomy, administration of corticosteroids, or low calcium diet. The P-C-P bond of the bisphosphonates is completely resistant to enzymatic hydrolysis. The bisphosphonates studied up to now, such as etidronate, clodronate, pamidronate, and alendronate, are absorbed, stored, and excreted unaltered. The intestinal absorption of the bisphosphonates is low, between 1% or less and 10% of the amount ingested. The newer bisphosphonates are at the lower end of the scale. The absorption diminishes when the compounds are given with food, especially in the presence of calcium. Bisphosphonates are rapidly cleared from plasma, 20%-80% being deposited in bone and the remainder excreted in the urine. In bone, they deposit at sites of mineralization as well as under the osteoclasts. In contrast to plasma, the half-life in bone is very long, partially as long as the half-life of the bone in which they are deposited. In humans, bisphosphonates are used successfully in diseases with increased bone turnover, such as Paget's disease, tumoural bone disease, as well as in osteoporosis. Various bisphosphonates, such as alendronate, clodronate, etidronate, ibandronate, pamidronate, and tiludronate, have been investigated in osteoporosis. All inhibit bone loss in postmenopausal women and increase bone mass. Furthermore, bisphosphonates are also effective in preventing bone loss both in corticosteroid-treated and in immobilized patients. The effect on the rate of fractures has recently been proven for alendronate. In humans, the adverse effects depend upon the compound and the amount given. For etidronate, practically the only adverse effect is an inhibition of mineralization. The aminoderivatives induce for a period of 2-3 days a syndrome with pyrexia, which shows a similitude with an acute phase reaction. The more potent compounds can induce gastrointestinal disturbances, sometimes oesophagitis, when given orally. Bisphosphonates are an important addition to the therapeutic possibilities in the prevention and treatment of osteoporosis. PMID: [PubMed - indexed for MEDLINE]
: Links. Bisphosphonates: preclinical aspects and use in osteoporosis. Fleisch HA. Department of Pathophysiology, University of Bern, Switzerland. The bisphosphonates are synthetic compounds characterized by a P-C-P bond. They have a strong affinity to calcium phosphates and hence to bone mineral. In vitro they inhibit both formation and dissolution of the latter. Many of the bisphosphonates inhibit bone resorption, the newest compounds being 10,000 times more active than etidronate, the first bisphosphonate described. The antiresorbing effect is cell mediated, partly by a direct action on the osteoclasts, partly through the osteoblasts, which produce an inhibitor of osteoclastic recruitment. When given in large amounts, some bisphosphonates can also inhibit normal and ectopic mineralization through a physical-chemical inhibition of crystal growth. In the growing rat the inhibition of resorption is accompanied by an increase in intestinal absorption and an increased balance of calcium. Bisphosphonates also prevent various types of experimental osteoporosis, such as after immobilization, ovariectomy, orchidectomy, administration of corticosteroids, or low calcium diet. The P-C-P bond of the bisphosphonates is completely resistant to enzymatic hydrolysis. The bisphosphonates studied up to now, such as etidronate, clodronate, pamidronate, and alendronate, are absorbed, stored, and excreted unaltered. The intestinal absorption of the bisphosphonates is low, between 1% or less and 10% of the amount ingested. The newer bisphosphonates are at the lower end of the scale. The absorption diminishes when the compounds are given with food, especially in the presence of calcium. Bisphosphonates are rapidly cleared from plasma, 20%-80% being deposited in bone and the remainder excreted in the urine. In bone, they deposit at sites of mineralization as well as under the osteoclasts. In contrast to plasma, the half-life in bone is very long, partially as long as the half-life of the bone in which they are deposited. In humans, bisphosphonates are used successfully in diseases with increased bone turnover, such as Paget s disease, tumoural bone disease, as well as in osteoporosis. Various bisphosphonates, such as alendronate, clodronate, etidronate, ibandronate, pamidronate, and tiludronate, have been investigated in osteoporosis. All inhibit bone loss in postmenopausal women and increase bone mass. Furthermore, bisphosphonates are also effective in preventing bone loss both in corticosteroid-treated and in immobilized patients. The effect on the rate of fractures has recently been proven for alendronate. In humans, the adverse effects depend upon the compound and the amount given. For etidronate, practically the only adverse effect is an inhibition of mineralization. The aminoderivatives induce for a period of 2-3 days a syndrome with pyrexia, which shows a similitude with an acute phase reaction. The more potent compounds can induce gastrointestinal disturbances, sometimes oesophagitis, when given orally. Bisphosphonates are an important addition to the therapeutic possibilities in the prevention and treatment of osteoporosis. PMID: [PubMed - indexed for MEDLINE]")
36
Uyarılar Bisfosfonatlar özofajit, özofagus ülserleri ve erozyonlarına neden olabilirler. Sabah aç karna ve bol su ile birlikte alınmalı, alındıktan sonra 30 dakika yiyecek ve su dışında içecek alınmamalı ve yatılmamalıdır. Günün başka bir saatinde alınacak ise, en az 2 saat önceden 2 saat sonraya kadar yiyecek ve su dışında içecek alınmamalıdır.

37
Endikasyonları Kontrendikasyonlar Osteoporoz
Paget Hastalığı (ilk tercih) Tümörle ilişkili osteolitik kemik hastalığı Renal fonksiyon bozukluğu Özofageal motilite bozuklukları Peptik ülser Gebelik UpToDate 14.1 Recommendations — We consider the bisphosphonates (alendronate or risedronate), and raloxifene as first-line treatments for prevention of osteoporosis, and bisphosphonates as first-line therapy for treatment of osteoporosis. We favor bisphosphonates over raloxifene for osteoporosis treatment because they increase bone mineral density more than raloxifene [83], although the clinical significance of this observation is unclear [29,84,85]. In addition, there are no data indicating that raloxifene prevents hip fractures. Estrogen therapy should no longer be used solely for the prevention of postmenopausal osteoporosis. Exceptions include women with persistent menopausal symptoms and those who cannot tolerate or do not adequately respond to the other drugs. As noted above, combination therapy can produce an additive response. (See "Postmenopausal hormone therapy: Benefits and risks" and see "Recommendations for postmenopausal hormone therapy"). Human recombinant PTH should be considered for patients at high risk for fracture who have not responded to oral bisphosphonate therapy (see "Parathyroid hormone" above).
 Tümörle ilişkili osteolitik kemik hastalığı. Renal fonksiyon bozukluğu. Özofageal motilite bozuklukları. Peptik ülser. Gebelik. UpToDate Recommendations — We consider the bisphosphonates (alendronate or risedronate), and raloxifene as first-line treatments for prevention of osteoporosis, and bisphosphonates as first-line therapy for treatment of osteoporosis. We favor bisphosphonates over raloxifene for osteoporosis treatment because they increase bone mineral density more than raloxifene [83], although the clinical significance of this observation is unclear [29,84,85]. In addition, there are no data indicating that raloxifene prevents hip fractures. Estrogen therapy should no longer be used solely for the prevention of postmenopausal osteoporosis. Exceptions include women with persistent menopausal symptoms and those who cannot tolerate or do not adequately respond to the other drugs. As noted above, combination therapy can produce an additive response. (See Postmenopausal hormone therapy: Benefits and risks and see Recommendations for postmenopausal hormone therapy ). Human recombinant PTH should be considered for patients at high risk for fracture who have not responded to oral bisphosphonate therapy (see Parathyroid hormone above).")
38
SERM Selektif Estrojen Reseptör Modülatörleri (Tamoksifen, Raloksifen)
NOLVADEX 10 mg, 30 tablet 24,91 YTL 10 mg, 250 tablet 198,07 YTL 20 mg, 30 tablet 39,48 YTL TADEX 10 mg, 30 tablet 14,55 YTL 10 mg, 250 tablet 60,67 YTL TAMOPLEX 20 mg, 30 tablet 21,51 YTL TAMOXIFEN 10 mg, 30 tablet 7,83 YTL 10 mg, 100 tablet 20,69 YTL 20 mg, 30 tablet 27,10 YTL 20 mg, 100 tablet 35,22 YTL TAMOXIFENO GADOR 10 mg, 30 tablet 14,55 YTL 10 mg, 250 tablet 64,85 YTL 20 mg, 30 tablet 20,99 YTL TAMOXIFEN TABLET-BP 10 mg, 30 tablet 13,99 YTL 20 mg, 30 tablet 18,11 YTL Menapozal kemik kaybına karşı koruyucu etkiye sahiptirler. Ancak kemik dansitesinde sağladıkları artış (2 yıllık tedavide %1,2) estrojen tedavisi ile sağlananın (2 yıllık tedavide %5-7) altındadır. INTRODUCTION – Two classes of drugs are available that act as selective estrogen receptor modulators (SERMs): tamoxifen and raloxifene and related drugs [1]. Both agents are competitive inhibitors of estrogen binding to estrogen receptors (ERs), and both have mixed agonist and antagonist activity, depending on the target tissue. These mixed activities have led to the redesignation of this class of compounds from "anti-estrogens" to SERMs. Both tamoxifen and raloxifene provide some protection against menopausal bone loss, presumably due to their partial agonist activity (show figure 1 and show figure 2) [3-6]. However, the increase in bone density (about 1.2 percent in the lumbar spine at two years) is substantially less than that seen with estrogen therapy (5 to 7 percent at two years). • Both agents lower serum total and low-density lipoprotein (LDL)-cholesterol concentrations (by 12 and 19 percent, respectively, in one report) although they do not increase serum HDL-cholesterol [5,7-9]. However, data confirming protection against cardiovascular disease for either drug, or for estrogen, are still controversial. (See "Use of selective estrogen receptor modulators in postmenopausal women"). • Tamoxifen's antagonist effect is particularly prominent with respect to breast cancer. Among women with ER-positive breast cancer, tamoxifen reduces the risk of recurrence and death when given as adjuvant therapy for early stage disease, and can provide palliation in those with metastatic disease [10,11]. However, as will be described below, some ER-positive breast cancers display primary resistance to tamoxifen, and all advanced breast cancers eventually become refractory to tamoxifen treatment (secondary resistance). Tamoxifen may also prevent the development of contralateral breast cancer, both in women with a prior diagnosis of breast cancer, and in those women at high risk of breast cancer (show figure 3) [8]. (See "Selective estrogen receptor modulators for the prevention of breast cancer"). Women patients and their physicians can roughly estimate the annual and lifetime risk of developing breast cancer using a simple algorithm based on the Gail model and available online ( [12]. (See "Risk assessment in women with an inherited predisposition to breast cancer-I", section on Empiric models to assess the risk of developing breast cancer). • Raloxifene may also protect against the development of breast cancer [5,6]. The National Surgical Adjuvant Breast and Bowel Project (NSABP) Study of Tamoxifen and Raloxifene (STAR trial, website is one of the largest breast cancer prevention studies to be ever conducted. This five year study aims to recruit 19,000 volunteers at more than 400 centers across the United States, Puerto Rico, and Canada. Accrual was reached in the summer of 2004, and early results are expected in 2006. • Both raloxifene and tamoxifen also induce hot flashes (an estrogen antagonist effect). Of interest, while tamoxifen clearly induces endometrial hyperplasia (an estrogen agonist effect) [13], and increases the risk of developing endometrial cancer (show figure 4) [8,10], raloxifene does not appear to have endometrioid agonistic effects. It does not predispose to endometrial cancer [5,6,14]. As a result of the increased risk of developing endometrial cancer with tamoxifen, the American College of Obstetricians and Gynecologists has developed recommendations for monitoring women on tamoxifen for this complication [15]. (See "Use of selective estrogen receptor modulators in postmenopausal women", section on Adverse effects). How tamoxifen and raloxifene can have both antagonist and agonist actions on the ER in different tissues will be reviewed here, beginning with an overview of the function of the ER. The emphasis will be on the effects of tamoxifen on breast cancer. Additional material on the physiology of estrogen and estrogen receptors is presented elsewhere. (See "Physiology of estrogen action"). THE ESTROGEN RECEPTOR (ER) – Most receptors of the steroid family, with the exception of the ER, are classically viewed as 'translocating receptors'. That is, they move from a principally cytoplasmic distribution in the absence of hormone to a predominantly nuclear localization in hormone stimulated cells. However, the ER appears to be predominantly nuclear both in the presence and absence of hormone. The ER operates as a ligand-dependent transcription factor; attachment of estrogen hormone to the ER's ligand-binding domain results in either direct binding of the ER to estrogen response elements (ERE) in the promoter of target genes, or to a protein-protein interaction with coactivators at their respective promoter sites [16-19]. Subsequently, the hormone-receptor complex is able to bind to estrogen-specific response elements that activate or repress expression of genes whose protein products are responsible for the physiologic actions of the hormone. The ER shares many structural features with other members of the nuclear receptor superfamily with six components or "domains", A to F (show figure 5) [17,18]. Estradiol and SERMs such as tamoxifen bind to the ligand-binding site in the E domain, which also mediates ER dimerization. The sequence-specific DNA binding function of the ER requires dimerization and resides in domain C. Domain D contains a nuclear localization signal. Regions that promote transcription activation functions are present in domains A/B (AF1) and E (AF2). (See "Physiology of estrogen action"). Corepressors and coactivators – On a molecular level, we are only beginning to understand how an individual SERM can act as an ER agonist in one tissue and as an antagonist in another. However, it is likely that the change in receptor conformation that follows binding of the ER by a SERM results in variable interactions with co-factors that are required for ER-mediated gene regulation. These nuclear proteins, termed coactivators and corepressors, are able to interpret the difference between binding of different ligands (eg, estrogen or tamoxifen) to the ER. Coactivators increase the transcriptional activity of the ER by promoting an interaction between the receptor and the transcriptional apparatus that provides the machinery for gene activation and subsequent mRNA transcription [18]. In contrast, corepressors restrain ER activity, maintaining the receptor in a protein/DNA complex that does not promote transcription [20]. ER coactivators are exquisitely sensitive to the conformational changes that occur in the ligand-binding domain (LBD) [21-27]. Unlike estrogens, tamoxifen distorts the LBD, generating an abnormal receptor conformation that disrupts coactivator binding outside the LBD [24,28-30]. Subsequently corepressor molecules are recruited to the ER, holding it in an inactive state [31]. The influence of the available coactivators and corepressors on the tissue specificity of tamoxifen, and the development of tamoxifen resistance is discussed below. Estrogen receptor beta: a second ER isoform – The above characteristics describe ER-alpha. A second isoform, ER-beta, has been described that is highly homologous to ER-alpha in both the DNA binding and ligand binding domains [32,33]. ER-beta binds estrogens with a similar affinity as ER-alpha, and activates the expression of genes containing estrogen response elements (ERE) in an estrogen-dependent manner [34]. In contrast to the hormone and DNA binding domains, ER-alpha and ER-beta are not homologous in the N terminal A/B (transactivation) domains. ER-beta does not contain a strong AF1 within its amino-terminus but, rather, contains a repressor domain that when removed, increases the overall transcriptional activity of the receptor. As a result, the transcriptional properties of ER-alpha and ER-beta are dissimilar. (See "Physiology of estrogen action"). Transcriptional activity of ER-beta in response to estrogen is dependent upon the cell type, promoter, and the nature of the ligand. Tamoxifen, which shows agonist activity in some tissues upon binding to ER-alpha, has no agonistic activity when it interacts with ER-beta. These differences appear to result from alterations in the amino-terminal A/B receptor domain, particularly AF1 [35]. Unlike ER-alpha (see below), ER-beta does not have a strong AF1 domain, and its AF2 domain appears to function independently within the receptor [36]. In addition, the partial agonist activity of tamoxifen that is manifest through ER-alpha can be completely abolished upon coexpression of ER-beta [36-38]. When coexpressed in tumor cells, ER-beta functions as a transdominant inhibitor of ER-alpha transcriptional activity at subsaturating hormone levels, and decreases overall cellular sensitivity to estradiol. The discovery of ER-beta has added further complexity to the molecular biology of the ER and its interaction with estrogens and SERMs in different tissues. Further study of these interactions, particularly using molecular methods such as gene expression profiling, may help explain the differential actions of tamoxifen and other SERMs on different tissues [39]. Currently, information on the clinical significance of ER-beta expression in breast cancer is limited. INTERACTION OF TAMOXIFEN WITH THE ER – A simple model of ER-alpha function is provided in Figure 6 (show figure 6). After estrogen or tamoxifen binds to the ligand-binding domain, the ER is released from heat shock protein (HSP)-90 and ER dimerization occurs. Sequence-specific DNA-binding to an ERE follows. In the presence of estrogen, mRNA transcription is promoted though AF2. In figure 6, tamoxifen-bound ERs are shown as inactive, since tamoxifen inhibits AF2 function in breast cancer cells. However, this simple paradigm provides no insight into the tissue-specific mixed agonist/antagonist actions of SERMs such as tamoxifen [17]. As a result, more complex models are required (show figure 7) [19]. (See "Overview of gene expression profiling and proteomics in clinical oncology-I"). INTERACTION OF RALOXIFENE WITH ESTROGEN RECEPTOR ALPHA – The newer SERMs such as raloxifene appear to have different tissue-specific effects from tamoxifen. Those factors that determine the variable ER agonist and antagonist activity of raloxifene are not fully defined but are under active study [39]. Like tamoxifen, raloxifene distorts the LBD of the ER, generating an abnormal receptor conformation that disrupts coactivator binding [38]. As a result, there is likely to be significant cross-resistance between tamoxifen and raloxifene, resulting in a relative lack of activity of raloxifene in tamoxifen-refractory breast cancer. The differing effects of raloxifene and tamoxifen on the uterus may be related to structural differences in the two compounds that influence the conformations of the ligand-receptor complexes, thereby determining which estrogen-responsive genes are modulated in specific tissues [65]. Alternatively, differences in tissue specificity compared to tamoxifen may be related to other unique aspects of the interaction of raloxifene with the ER. As an example, the human transforming growth factor-beta3 gene is activated by the ER in the presence of estrogen metabolites or estrogen antagonists such as raloxifene [66]. Activation is mediated by a polypurine sequence, termed the raloxifene response element (RFE), and does not require the DNA binding domain of the estrogen receptor. Interaction of the estrogen receptor with the RFE appears to require a cellular adapter protein. The observation that individual estrogens and antagonists can modulate multiple DNA response elements may explain the tissue-selective estrogen agonist or antagonist activity of SERMs such as raloxifene. Raloksifen EVİSTA 60 mg, 28 tablet 53,91 YTL
 estrojen tedavisi ile sağlananın (2 yıllık tedavide %5-7) altındadır. INTRODUCTION – Two classes of drugs are available that act as selective estrogen receptor modulators (SERMs): tamoxifen and raloxifene and related drugs [1]. Both agents are competitive inhibitors of estrogen binding to estrogen receptors (ERs), and both have mixed agonist and antagonist activity, depending on the target tissue. These mixed activities have led to the redesignation of this class of compounds from anti-estrogens to SERMs. Both tamoxifen and raloxifene provide some protection against menopausal bone loss, presumably due to their partial agonist activity (show figure 1 and show figure 2) [3-6]. However, the increase in bone density (about 1.2 percent in the lumbar spine at two years) is substantially less than that seen with estrogen therapy (5 to 7 percent at two years). • Both agents lower serum total and low-density lipoprotein (LDL)-cholesterol concentrations (by 12 and 19 percent, respectively, in one report) although they do not increase serum HDL-cholesterol [5,7-9]. However, data confirming protection against cardiovascular disease for either drug, or for estrogen, are still controversial. (See Use of selective estrogen receptor modulators in postmenopausal women ). • Tamoxifen s antagonist effect is particularly prominent with respect to breast cancer. Among women with ER-positive breast cancer, tamoxifen reduces the risk of recurrence and death when given as adjuvant therapy for early stage disease, and can provide palliation in those with metastatic disease [10,11]. However, as will be described below, some ER-positive breast cancers display primary resistance to tamoxifen, and all advanced breast cancers eventually become refractory to tamoxifen treatment (secondary resistance). Tamoxifen may also prevent the development of contralateral breast cancer, both in women with a prior diagnosis of breast cancer, and in those women at high risk of breast cancer (show figure 3) [8]. (See Selective estrogen receptor modulators for the prevention of breast cancer ). Women patients and their physicians can roughly estimate the annual and lifetime risk of developing breast cancer using a simple algorithm based on the Gail model and available online ( [12]. (See Risk assessment in women with an inherited predisposition to breast cancer-I , section on Empiric models to assess the risk of developing breast cancer). • Raloxifene may also protect against the development of breast cancer [5,6]. The National Surgical Adjuvant Breast and Bowel Project (NSABP) Study of Tamoxifen and Raloxifene (STAR trial, website is one of the largest breast cancer prevention studies to be ever conducted. This five year study aims to recruit 19,000 volunteers at more than 400 centers across the United States, Puerto Rico, and Canada. Accrual was reached in the summer of 2004, and early results are expected in • Both raloxifene and tamoxifen also induce hot flashes (an estrogen antagonist effect). Of interest, while tamoxifen clearly induces endometrial hyperplasia (an estrogen agonist effect) [13], and increases the risk of developing endometrial cancer (show figure 4) [8,10], raloxifene does not appear to have endometrioid agonistic effects. It does not predispose to endometrial cancer [5,6,14]. As a result of the increased risk of developing endometrial cancer with tamoxifen, the American College of Obstetricians and Gynecologists has developed recommendations for monitoring women on tamoxifen for this complication [15]. (See Use of selective estrogen receptor modulators in postmenopausal women , section on Adverse effects). How tamoxifen and raloxifene can have both antagonist and agonist actions on the ER in different tissues will be reviewed here, beginning with an overview of the function of the ER. The emphasis will be on the effects of tamoxifen on breast cancer. Additional material on the physiology of estrogen and estrogen receptors is presented elsewhere. (See Physiology of estrogen action ). THE ESTROGEN RECEPTOR (ER) – Most receptors of the steroid family, with the exception of the ER, are classically viewed as translocating receptors . That is, they move from a principally cytoplasmic distribution in the absence of hormone to a predominantly nuclear localization in hormone stimulated cells. However, the ER appears to be predominantly nuclear both in the presence and absence of hormone. The ER operates as a ligand-dependent transcription factor; attachment of estrogen hormone to the ER s ligand-binding domain results in either direct binding of the ER to estrogen response elements (ERE) in the promoter of target genes, or to a protein-protein interaction with coactivators at their respective promoter sites [16-19]. Subsequently, the hormone-receptor complex is able to bind to estrogen-specific response elements that activate or repress expression of genes whose protein products are responsible for the physiologic actions of the hormone. The ER shares many structural features with other members of the nuclear receptor superfamily with six components or domains , A to F (show figure 5) [17,18]. Estradiol and SERMs such as tamoxifen bind to the ligand-binding site in the E domain, which also mediates ER dimerization. The sequence-specific DNA binding function of the ER requires dimerization and resides in domain C. Domain D contains a nuclear localization signal. Regions that promote transcription activation functions are present in domains A/B (AF1) and E (AF2). (See Physiology of estrogen action ). Corepressors and coactivators – On a molecular level, we are only beginning to understand how an individual SERM can act as an ER agonist in one tissue and as an antagonist in another. However, it is likely that the change in receptor conformation that follows binding of the ER by a SERM results in variable interactions with co-factors that are required for ER-mediated gene regulation. These nuclear proteins, termed coactivators and corepressors, are able to interpret the difference between binding of different ligands (eg, estrogen or tamoxifen) to the ER. Coactivators increase the transcriptional activity of the ER by promoting an interaction between the receptor and the transcriptional apparatus that provides the machinery for gene activation and subsequent mRNA transcription [18]. In contrast, corepressors restrain ER activity, maintaining the receptor in a protein/DNA complex that does not promote transcription [20]. ER coactivators are exquisitely sensitive to the conformational changes that occur in the ligand-binding domain (LBD) [21-27]. Unlike estrogens, tamoxifen distorts the LBD, generating an abnormal receptor conformation that disrupts coactivator binding outside the LBD [24,28-30]. Subsequently corepressor molecules are recruited to the ER, holding it in an inactive state [31]. The influence of the available coactivators and corepressors on the tissue specificity of tamoxifen, and the development of tamoxifen resistance is discussed below. Estrogen receptor beta: a second ER isoform – The above characteristics describe ER-alpha. A second isoform, ER-beta, has been described that is highly homologous to ER-alpha in both the DNA binding and ligand binding domains [32,33]. ER-beta binds estrogens with a similar affinity as ER-alpha, and activates the expression of genes containing estrogen response elements (ERE) in an estrogen-dependent manner [34]. In contrast to the hormone and DNA binding domains, ER-alpha and ER-beta are not homologous in the N terminal A/B (transactivation) domains. ER-beta does not contain a strong AF1 within its amino-terminus but, rather, contains a repressor domain that when removed, increases the overall transcriptional activity of the receptor. As a result, the transcriptional properties of ER-alpha and ER-beta are dissimilar. (See Physiology of estrogen action ). Transcriptional activity of ER-beta in response to estrogen is dependent upon the cell type, promoter, and the nature of the ligand. Tamoxifen, which shows agonist activity in some tissues upon binding to ER-alpha, has no agonistic activity when it interacts with ER-beta. These differences appear to result from alterations in the amino-terminal A/B receptor domain, particularly AF1 [35]. Unlike ER-alpha (see below), ER-beta does not have a strong AF1 domain, and its AF2 domain appears to function independently within the receptor [36]. In addition, the partial agonist activity of tamoxifen that is manifest through ER-alpha can be completely abolished upon coexpression of ER-beta [36-38]. When coexpressed in tumor cells, ER-beta functions as a transdominant inhibitor of ER-alpha transcriptional activity at subsaturating hormone levels, and decreases overall cellular sensitivity to estradiol. The discovery of ER-beta has added further complexity to the molecular biology of the ER and its interaction with estrogens and SERMs in different tissues. Further study of these interactions, particularly using molecular methods such as gene expression profiling, may help explain the differential actions of tamoxifen and other SERMs on different tissues [39]. Currently, information on the clinical significance of ER-beta expression in breast cancer is limited. INTERACTION OF TAMOXIFEN WITH THE ER – A simple model of ER-alpha function is provided in Figure 6 (show figure 6). After estrogen or tamoxifen binds to the ligand-binding domain, the ER is released from heat shock protein (HSP)-90 and ER dimerization occurs. Sequence-specific DNA-binding to an ERE follows. In the presence of estrogen, mRNA transcription is promoted though AF2. In figure 6, tamoxifen-bound ERs are shown as inactive, since tamoxifen inhibits AF2 function in breast cancer cells. However, this simple paradigm provides no insight into the tissue-specific mixed agonist/antagonist actions of SERMs such as tamoxifen [17]. As a result, more complex models are required (show figure 7) [19]. (See Overview of gene expression profiling and proteomics in clinical oncology-I ). INTERACTION OF RALOXIFENE WITH ESTROGEN RECEPTOR ALPHA – The newer SERMs such as raloxifene appear to have different tissue-specific effects from tamoxifen. Those factors that determine the variable ER agonist and antagonist activity of raloxifene are not fully defined but are under active study [39]. Like tamoxifen, raloxifene distorts the LBD of the ER, generating an abnormal receptor conformation that disrupts coactivator binding [38]. As a result, there is likely to be significant cross-resistance between tamoxifen and raloxifene, resulting in a relative lack of activity of raloxifene in tamoxifen-refractory breast cancer. The differing effects of raloxifene and tamoxifen on the uterus may be related to structural differences in the two compounds that influence the conformations of the ligand-receptor complexes, thereby determining which estrogen-responsive genes are modulated in specific tissues [65]. Alternatively, differences in tissue specificity compared to tamoxifen may be related to other unique aspects of the interaction of raloxifene with the ER. As an example, the human transforming growth factor-beta3 gene is activated by the ER in the presence of estrogen metabolites or estrogen antagonists such as raloxifene [66]. Activation is mediated by a polypurine sequence, termed the raloxifene response element (RFE), and does not require the DNA binding domain of the estrogen receptor. Interaction of the estrogen receptor with the RFE appears to require a cellular adapter protein. The observation that individual estrogens and antagonists can modulate multiple DNA response elements may explain the tissue-selective estrogen agonist or antagonist activity of SERMs such as raloxifene. Raloksifen. EVİSTA 60 mg, 28 tablet 53,91 YTL.")
39
STRONSİYUM RANELAT Kemik oluşumunu uyarır, kemik rezorpsiyonunu azaltır. Postmenopozal osteoporoz tedavisi için onaylanmıştır. PROTELOS 2 g/saşe, 28 saşe 78,30 YTL INTRODUCTION – Two classes of drugs are available that act as selective estrogen receptor modulators (SERMs): tamoxifen and raloxifene and related drugs [1]. Both agents are competitive inhibitors of estrogen binding to estrogen receptors (ERs), and both have mixed agonist and antagonist activity, depending on the target tissue. These mixed activities have led to the redesignation of this class of compounds from "anti-estrogens" to SERMs. Both tamoxifen and raloxifene provide some protection against menopausal bone loss, presumably due to their partial agonist activity (show figure 1 and show figure 2) [3-6]. However, the increase in bone density (about 1.2 percent in the lumbar spine at two years) is substantially less than that seen with estrogen therapy (5 to 7 percent at two years). • Both agents lower serum total and low-density lipoprotein (LDL)-cholesterol concentrations (by 12 and 19 percent, respectively, in one report) although they do not increase serum HDL-cholesterol [5,7-9]. However, data confirming protection against cardiovascular disease for either drug, or for estrogen, are still controversial. (See "Use of selective estrogen receptor modulators in postmenopausal women"). • Tamoxifen's antagonist effect is particularly prominent with respect to breast cancer. Among women with ER-positive breast cancer, tamoxifen reduces the risk of recurrence and death when given as adjuvant therapy for early stage disease, and can provide palliation in those with metastatic disease [10,11]. However, as will be described below, some ER-positive breast cancers display primary resistance to tamoxifen, and all advanced breast cancers eventually become refractory to tamoxifen treatment (secondary resistance). Tamoxifen may also prevent the development of contralateral breast cancer, both in women with a prior diagnosis of breast cancer, and in those women at high risk of breast cancer (show figure 3) [8]. (See "Selective estrogen receptor modulators for the prevention of breast cancer"). Women patients and their physicians can roughly estimate the annual and lifetime risk of developing breast cancer using a simple algorithm based on the Gail model and available online ( [12]. (See "Risk assessment in women with an inherited predisposition to breast cancer-I", section on Empiric models to assess the risk of developing breast cancer). • Raloxifene may also protect against the development of breast cancer [5,6]. The National Surgical Adjuvant Breast and Bowel Project (NSABP) Study of Tamoxifen and Raloxifene (STAR trial, website is one of the largest breast cancer prevention studies to be ever conducted. This five year study aims to recruit 19,000 volunteers at more than 400 centers across the United States, Puerto Rico, and Canada. Accrual was reached in the summer of 2004, and early results are expected in 2006. • Both raloxifene and tamoxifen also induce hot flashes (an estrogen antagonist effect). Of interest, while tamoxifen clearly induces endometrial hyperplasia (an estrogen agonist effect) [13], and increases the risk of developing endometrial cancer (show figure 4) [8,10], raloxifene does not appear to have endometrioid agonistic effects. It does not predispose to endometrial cancer [5,6,14]. As a result of the increased risk of developing endometrial cancer with tamoxifen, the American College of Obstetricians and Gynecologists has developed recommendations for monitoring women on tamoxifen for this complication [15]. (See "Use of selective estrogen receptor modulators in postmenopausal women", section on Adverse effects). How tamoxifen and raloxifene can have both antagonist and agonist actions on the ER in different tissues will be reviewed here, beginning with an overview of the function of the ER. The emphasis will be on the effects of tamoxifen on breast cancer. Additional material on the physiology of estrogen and estrogen receptors is presented elsewhere. (See "Physiology of estrogen action"). THE ESTROGEN RECEPTOR (ER) – Most receptors of the steroid family, with the exception of the ER, are classically viewed as 'translocating receptors'. That is, they move from a principally cytoplasmic distribution in the absence of hormone to a predominantly nuclear localization in hormone stimulated cells. However, the ER appears to be predominantly nuclear both in the presence and absence of hormone. The ER operates as a ligand-dependent transcription factor; attachment of estrogen hormone to the ER's ligand-binding domain results in either direct binding of the ER to estrogen response elements (ERE) in the promoter of target genes, or to a protein-protein interaction with coactivators at their respective promoter sites [16-19]. Subsequently, the hormone-receptor complex is able to bind to estrogen-specific response elements that activate or repress expression of genes whose protein products are responsible for the physiologic actions of the hormone. The ER shares many structural features with other members of the nuclear receptor superfamily with six components or "domains", A to F (show figure 5) [17,18]. Estradiol and SERMs such as tamoxifen bind to the ligand-binding site in the E domain, which also mediates ER dimerization. The sequence-specific DNA binding function of the ER requires dimerization and resides in domain C. Domain D contains a nuclear localization signal. Regions that promote transcription activation functions are present in domains A/B (AF1) and E (AF2). (See "Physiology of estrogen action"). Corepressors and coactivators – On a molecular level, we are only beginning to understand how an individual SERM can act as an ER agonist in one tissue and as an antagonist in another. However, it is likely that the change in receptor conformation that follows binding of the ER by a SERM results in variable interactions with co-factors that are required for ER-mediated gene regulation. These nuclear proteins, termed coactivators and corepressors, are able to interpret the difference between binding of different ligands (eg, estrogen or tamoxifen) to the ER. Coactivators increase the transcriptional activity of the ER by promoting an interaction between the receptor and the transcriptional apparatus that provides the machinery for gene activation and subsequent mRNA transcription [18]. In contrast, corepressors restrain ER activity, maintaining the receptor in a protein/DNA complex that does not promote transcription [20]. ER coactivators are exquisitely sensitive to the conformational changes that occur in the ligand-binding domain (LBD) [21-27]. Unlike estrogens, tamoxifen distorts the LBD, generating an abnormal receptor conformation that disrupts coactivator binding outside the LBD [24,28-30]. Subsequently corepressor molecules are recruited to the ER, holding it in an inactive state [31]. The influence of the available coactivators and corepressors on the tissue specificity of tamoxifen, and the development of tamoxifen resistance is discussed below. Estrogen receptor beta: a second ER isoform – The above characteristics describe ER-alpha. A second isoform, ER-beta, has been described that is highly homologous to ER-alpha in both the DNA binding and ligand binding domains [32,33]. ER-beta binds estrogens with a similar affinity as ER-alpha, and activates the expression of genes containing estrogen response elements (ERE) in an estrogen-dependent manner [34]. In contrast to the hormone and DNA binding domains, ER-alpha and ER-beta are not homologous in the N terminal A/B (transactivation) domains. ER-beta does not contain a strong AF1 within its amino-terminus but, rather, contains a repressor domain that when removed, increases the overall transcriptional activity of the receptor. As a result, the transcriptional properties of ER-alpha and ER-beta are dissimilar. (See "Physiology of estrogen action"). Transcriptional activity of ER-beta in response to estrogen is dependent upon the cell type, promoter, and the nature of the ligand. Tamoxifen, which shows agonist activity in some tissues upon binding to ER-alpha, has no agonistic activity when it interacts with ER-beta. These differences appear to result from alterations in the amino-terminal A/B receptor domain, particularly AF1 [35]. Unlike ER-alpha (see below), ER-beta does not have a strong AF1 domain, and its AF2 domain appears to function independently within the receptor [36]. In addition, the partial agonist activity of tamoxifen that is manifest through ER-alpha can be completely abolished upon coexpression of ER-beta [36-38]. When coexpressed in tumor cells, ER-beta functions as a transdominant inhibitor of ER-alpha transcriptional activity at subsaturating hormone levels, and decreases overall cellular sensitivity to estradiol. The discovery of ER-beta has added further complexity to the molecular biology of the ER and its interaction with estrogens and SERMs in different tissues. Further study of these interactions, particularly using molecular methods such as gene expression profiling, may help explain the differential actions of tamoxifen and other SERMs on different tissues [39]. Currently, information on the clinical significance of ER-beta expression in breast cancer is limited. INTERACTION OF TAMOXIFEN WITH THE ER – A simple model of ER-alpha function is provided in Figure 6 (show figure 6). After estrogen or tamoxifen binds to the ligand-binding domain, the ER is released from heat shock protein (HSP)-90 and ER dimerization occurs. Sequence-specific DNA-binding to an ERE follows. In the presence of estrogen, mRNA transcription is promoted though AF2. In figure 6, tamoxifen-bound ERs are shown as inactive, since tamoxifen inhibits AF2 function in breast cancer cells. However, this simple paradigm provides no insight into the tissue-specific mixed agonist/antagonist actions of SERMs such as tamoxifen [17]. As a result, more complex models are required (show figure 7) [19]. (See "Overview of gene expression profiling and proteomics in clinical oncology-I"). INTERACTION OF RALOXIFENE WITH ESTROGEN RECEPTOR ALPHA – The newer SERMs such as raloxifene appear to have different tissue-specific effects from tamoxifen. Those factors that determine the variable ER agonist and antagonist activity of raloxifene are not fully defined but are under active study [39]. Like tamoxifen, raloxifene distorts the LBD of the ER, generating an abnormal receptor conformation that disrupts coactivator binding [38]. As a result, there is likely to be significant cross-resistance between tamoxifen and raloxifene, resulting in a relative lack of activity of raloxifene in tamoxifen-refractory breast cancer. The differing effects of raloxifene and tamoxifen on the uterus may be related to structural differences in the two compounds that influence the conformations of the ligand-receptor complexes, thereby determining which estrogen-responsive genes are modulated in specific tissues [65]. Alternatively, differences in tissue specificity compared to tamoxifen may be related to other unique aspects of the interaction of raloxifene with the ER. As an example, the human transforming growth factor-beta3 gene is activated by the ER in the presence of estrogen metabolites or estrogen antagonists such as raloxifene [66]. Activation is mediated by a polypurine sequence, termed the raloxifene response element (RFE), and does not require the DNA binding domain of the estrogen receptor. Interaction of the estrogen receptor with the RFE appears to require a cellular adapter protein. The observation that individual estrogens and antagonists can modulate multiple DNA response elements may explain the tissue-selective estrogen agonist or antagonist activity of SERMs such as raloxifene.
: tamoxifen and raloxifene and related drugs [1]. Both agents are competitive inhibitors of estrogen binding to estrogen receptors (ERs), and both have mixed agonist and antagonist activity, depending on the target tissue. These mixed activities have led to the redesignation of this class of compounds from anti-estrogens to SERMs. Both tamoxifen and raloxifene provide some protection against menopausal bone loss, presumably due to their partial agonist activity (show figure 1 and show figure 2) [3-6]. However, the increase in bone density (about 1.2 percent in the lumbar spine at two years) is substantially less than that seen with estrogen therapy (5 to 7 percent at two years). • Both agents lower serum total and low-density lipoprotein (LDL)-cholesterol concentrations (by 12 and 19 percent, respectively, in one report) although they do not increase serum HDL-cholesterol [5,7-9]. However, data confirming protection against cardiovascular disease for either drug, or for estrogen, are still controversial. (See Use of selective estrogen receptor modulators in postmenopausal women ). • Tamoxifen s antagonist effect is particularly prominent with respect to breast cancer. Among women with ER-positive breast cancer, tamoxifen reduces the risk of recurrence and death when given as adjuvant therapy for early stage disease, and can provide palliation in those with metastatic disease [10,11]. However, as will be described below, some ER-positive breast cancers display primary resistance to tamoxifen, and all advanced breast cancers eventually become refractory to tamoxifen treatment (secondary resistance). Tamoxifen may also prevent the development of contralateral breast cancer, both in women with a prior diagnosis of breast cancer, and in those women at high risk of breast cancer (show figure 3) [8]. (See Selective estrogen receptor modulators for the prevention of breast cancer ). Women patients and their physicians can roughly estimate the annual and lifetime risk of developing breast cancer using a simple algorithm based on the Gail model and available online ( [12]. (See Risk assessment in women with an inherited predisposition to breast cancer-I , section on Empiric models to assess the risk of developing breast cancer). • Raloxifene may also protect against the development of breast cancer [5,6]. The National Surgical Adjuvant Breast and Bowel Project (NSABP) Study of Tamoxifen and Raloxifene (STAR trial, website is one of the largest breast cancer prevention studies to be ever conducted. This five year study aims to recruit 19,000 volunteers at more than 400 centers across the United States, Puerto Rico, and Canada. Accrual was reached in the summer of 2004, and early results are expected in • Both raloxifene and tamoxifen also induce hot flashes (an estrogen antagonist effect). Of interest, while tamoxifen clearly induces endometrial hyperplasia (an estrogen agonist effect) [13], and increases the risk of developing endometrial cancer (show figure 4) [8,10], raloxifene does not appear to have endometrioid agonistic effects. It does not predispose to endometrial cancer [5,6,14]. As a result of the increased risk of developing endometrial cancer with tamoxifen, the American College of Obstetricians and Gynecologists has developed recommendations for monitoring women on tamoxifen for this complication [15]. (See Use of selective estrogen receptor modulators in postmenopausal women , section on Adverse effects). How tamoxifen and raloxifene can have both antagonist and agonist actions on the ER in different tissues will be reviewed here, beginning with an overview of the function of the ER. The emphasis will be on the effects of tamoxifen on breast cancer. Additional material on the physiology of estrogen and estrogen receptors is presented elsewhere. (See Physiology of estrogen action ). THE ESTROGEN RECEPTOR (ER) – Most receptors of the steroid family, with the exception of the ER, are classically viewed as translocating receptors . That is, they move from a principally cytoplasmic distribution in the absence of hormone to a predominantly nuclear localization in hormone stimulated cells. However, the ER appears to be predominantly nuclear both in the presence and absence of hormone. The ER operates as a ligand-dependent transcription factor; attachment of estrogen hormone to the ER s ligand-binding domain results in either direct binding of the ER to estrogen response elements (ERE) in the promoter of target genes, or to a protein-protein interaction with coactivators at their respective promoter sites [16-19]. Subsequently, the hormone-receptor complex is able to bind to estrogen-specific response elements that activate or repress expression of genes whose protein products are responsible for the physiologic actions of the hormone. The ER shares many structural features with other members of the nuclear receptor superfamily with six components or domains , A to F (show figure 5) [17,18]. Estradiol and SERMs such as tamoxifen bind to the ligand-binding site in the E domain, which also mediates ER dimerization. The sequence-specific DNA binding function of the ER requires dimerization and resides in domain C. Domain D contains a nuclear localization signal. Regions that promote transcription activation functions are present in domains A/B (AF1) and E (AF2). (See Physiology of estrogen action ). Corepressors and coactivators – On a molecular level, we are only beginning to understand how an individual SERM can act as an ER agonist in one tissue and as an antagonist in another. However, it is likely that the change in receptor conformation that follows binding of the ER by a SERM results in variable interactions with co-factors that are required for ER-mediated gene regulation. These nuclear proteins, termed coactivators and corepressors, are able to interpret the difference between binding of different ligands (eg, estrogen or tamoxifen) to the ER. Coactivators increase the transcriptional activity of the ER by promoting an interaction between the receptor and the transcriptional apparatus that provides the machinery for gene activation and subsequent mRNA transcription [18]. In contrast, corepressors restrain ER activity, maintaining the receptor in a protein/DNA complex that does not promote transcription [20]. ER coactivators are exquisitely sensitive to the conformational changes that occur in the ligand-binding domain (LBD) [21-27]. Unlike estrogens, tamoxifen distorts the LBD, generating an abnormal receptor conformation that disrupts coactivator binding outside the LBD [24,28-30]. Subsequently corepressor molecules are recruited to the ER, holding it in an inactive state [31]. The influence of the available coactivators and corepressors on the tissue specificity of tamoxifen, and the development of tamoxifen resistance is discussed below. Estrogen receptor beta: a second ER isoform – The above characteristics describe ER-alpha. A second isoform, ER-beta, has been described that is highly homologous to ER-alpha in both the DNA binding and ligand binding domains [32,33]. ER-beta binds estrogens with a similar affinity as ER-alpha, and activates the expression of genes containing estrogen response elements (ERE) in an estrogen-dependent manner [34]. In contrast to the hormone and DNA binding domains, ER-alpha and ER-beta are not homologous in the N terminal A/B (transactivation) domains. ER-beta does not contain a strong AF1 within its amino-terminus but, rather, contains a repressor domain that when removed, increases the overall transcriptional activity of the receptor. As a result, the transcriptional properties of ER-alpha and ER-beta are dissimilar. (See Physiology of estrogen action ). Transcriptional activity of ER-beta in response to estrogen is dependent upon the cell type, promoter, and the nature of the ligand. Tamoxifen, which shows agonist activity in some tissues upon binding to ER-alpha, has no agonistic activity when it interacts with ER-beta. These differences appear to result from alterations in the amino-terminal A/B receptor domain, particularly AF1 [35]. Unlike ER-alpha (see below), ER-beta does not have a strong AF1 domain, and its AF2 domain appears to function independently within the receptor [36]. In addition, the partial agonist activity of tamoxifen that is manifest through ER-alpha can be completely abolished upon coexpression of ER-beta [36-38]. When coexpressed in tumor cells, ER-beta functions as a transdominant inhibitor of ER-alpha transcriptional activity at subsaturating hormone levels, and decreases overall cellular sensitivity to estradiol. The discovery of ER-beta has added further complexity to the molecular biology of the ER and its interaction with estrogens and SERMs in different tissues. Further study of these interactions, particularly using molecular methods such as gene expression profiling, may help explain the differential actions of tamoxifen and other SERMs on different tissues [39]. Currently, information on the clinical significance of ER-beta expression in breast cancer is limited. INTERACTION OF TAMOXIFEN WITH THE ER – A simple model of ER-alpha function is provided in Figure 6 (show figure 6). After estrogen or tamoxifen binds to the ligand-binding domain, the ER is released from heat shock protein (HSP)-90 and ER dimerization occurs. Sequence-specific DNA-binding to an ERE follows. In the presence of estrogen, mRNA transcription is promoted though AF2. In figure 6, tamoxifen-bound ERs are shown as inactive, since tamoxifen inhibits AF2 function in breast cancer cells. However, this simple paradigm provides no insight into the tissue-specific mixed agonist/antagonist actions of SERMs such as tamoxifen [17]. As a result, more complex models are required (show figure 7) [19]. (See Overview of gene expression profiling and proteomics in clinical oncology-I ). INTERACTION OF RALOXIFENE WITH ESTROGEN RECEPTOR ALPHA – The newer SERMs such as raloxifene appear to have different tissue-specific effects from tamoxifen. Those factors that determine the variable ER agonist and antagonist activity of raloxifene are not fully defined but are under active study [39]. Like tamoxifen, raloxifene distorts the LBD of the ER, generating an abnormal receptor conformation that disrupts coactivator binding [38]. As a result, there is likely to be significant cross-resistance between tamoxifen and raloxifene, resulting in a relative lack of activity of raloxifene in tamoxifen-refractory breast cancer. The differing effects of raloxifene and tamoxifen on the uterus may be related to structural differences in the two compounds that influence the conformations of the ligand-receptor complexes, thereby determining which estrogen-responsive genes are modulated in specific tissues [65]. Alternatively, differences in tissue specificity compared to tamoxifen may be related to other unique aspects of the interaction of raloxifene with the ER. As an example, the human transforming growth factor-beta3 gene is activated by the ER in the presence of estrogen metabolites or estrogen antagonists such as raloxifene [66]. Activation is mediated by a polypurine sequence, termed the raloxifene response element (RFE), and does not require the DNA binding domain of the estrogen receptor. Interaction of the estrogen receptor with the RFE appears to require a cellular adapter protein. The observation that individual estrogens and antagonists can modulate multiple DNA response elements may explain the tissue-selective estrogen agonist or antagonist activity of SERMs such as raloxifene.")
40
Aşağıdaki ilaçlardan hangisinin vücutta kalış süresi en uzundur
Aşağıdaki ilaçlardan hangisinin vücutta kalış süresi en uzundur? A) Gabapentin B) Glipizid C) Finasterid D) Alendronat E) Penisilin G (Cevap D) 2004 Eylül
 Gabapentin B) Glipizid C) Finasterid D) Alendronat E) Penisilin G (Cevap D) 2004 Eylül.")
Benzer bir sunumlar

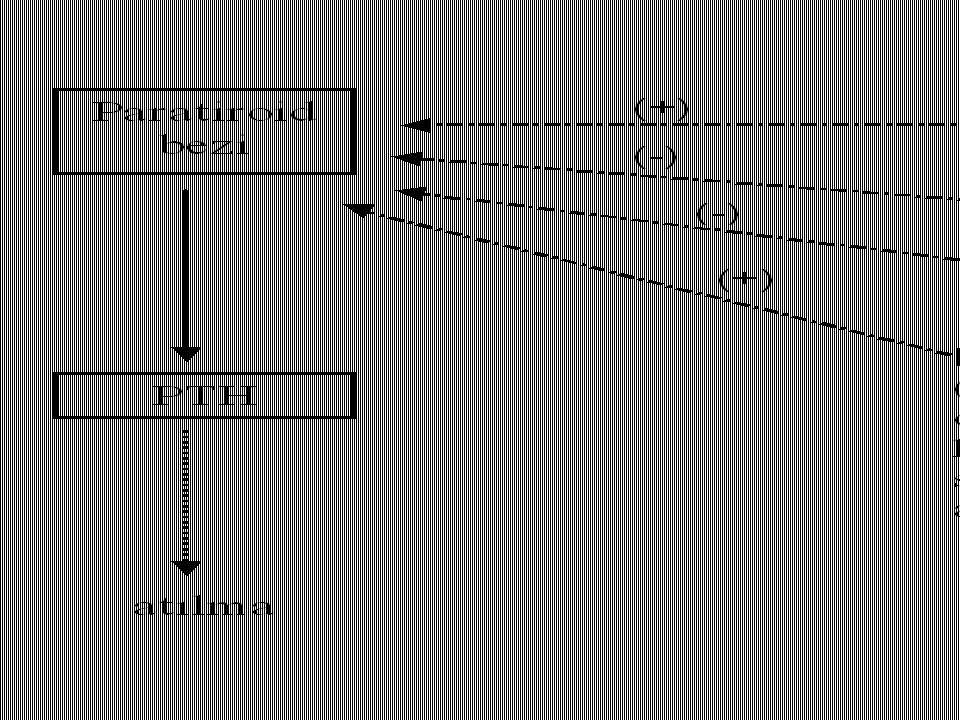
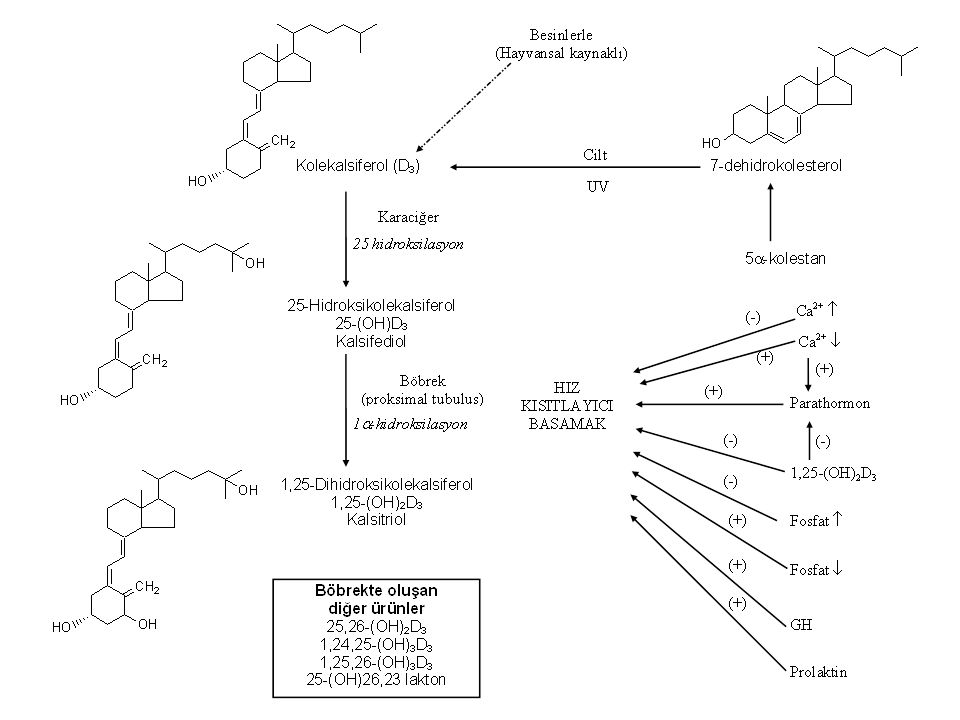













>")





