Sunuyu indir
1
BİYOYARARLANIM ve BİYOEŞDEĞERLİK
ESKİŞEHİR BİYOYARARLANIM ve BİYOEŞDEĞERLİK Anadolu Universitesi Eczacılık Fakültesi Farmasötik Teknoloji Anabilim Dalı

2
DERSİN İÇERİĞİ GİRİŞ Farmakokinetik Çalışmalar
Farmakodinamik Çalışmalar Klinik Çalışmalar İn vitro Çözünme Çalışmaları

3
BİYOYARARLANIM TANIM:
FDA: the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action Etkin maddenin veya onun terapötik molekül kısmının farmasötik şekilden absorbe edilerek sistemik dolaşıma geçme ve böylece vücuttaki etki yerinde veya onu yansıtan biyolojik sıvılarda (genellikle serum veya plazmada) var olma hızı ve derecesi
 var olma hızı ve derecesi.")
4
IV yoldan uygulanmayan bir ilaç için en önemli sorun kan dolaşımına ne kadarının geçtiği ve etki bölgesine ne kadarının ulaştığı Genellikle biyoyararlanımdan kasıt oral uygulama sonrasında GIS’den absorbe edilen ilacın biyoyararlanımı Uygulama; çözelti, süspansiyon, tablet, kapsül, toz gibi herhangi bir dozaj şekli ile yapılabilir IM enjeksiyon, transdermal yamalar, merhemler gibi topik uygulanan preparatların da sistemik dolaşıma geçebilmeleri için emilmeleri gerektiği için bu tip preparatların da BY’den bahsetmek mümkündür Bu sistemlerin tümü dolaşım sistemi dışından uygulama olarak kabul edilir BY’nin %100 kabul edildiği tek yol IV enjeksiyondur Uygulanan dozun tümü sistemik dolaşıma geçer

5
kan derişimi-zaman eğrisi kan derişimi-zaman eğrisi
1- Mutlak BY (Absolute) 2- Bağıl BY(Relative) 1- ilacın tamamen kana geçtiği bir yolla verilmesi ile elde edilen kan derişimi-zaman eğrisi ve AUC %100 kabul edilir = referans olarak kullanılır 2- Kana geçişin tam olduğu kabul edilen bir preparat ile BY karşılaştırılması kan derişimi-zaman eğrisi ve AUC
 2- Bağıl BY(Relative) 1- ilacın tamamen kana geçtiği bir yolla verilmesi ile elde edilen. kan derişimi-zaman eğrisi. ve. AUC. %100 kabul edilir = referans olarak kullanılır. 2- Kana geçişin tam olduğu kabul edilen bir preparat ile BY karşılaştırılması. kan derişimi-zaman eğrisi. ve. AUC.")
6
BİYOYARARLANIMIN ÖNEMİ
Farklı firmalar tarafından üretilen aynı etkin maddeyi taşıyan eşdeğer olduğu varsayılan ilaçların bir hastadan diğerine farklı terapötik etki göstermesinde biyoyararlanım anahtar rol oynamaktadır. Biyoyararlanım farkı, aynı firmada üretilen, aynı ilacın değişik serileri arasında da görülebilir

7
Biyoyararlanım çalışmalarında, dozaj şeklinden in vivo koşullarda serbest hale geçerek kan dolaşımına değişmeden geçen etkin maddenin oranı önemli Bu orana efektif veya biyolojik olarak yararlı doz da denir İlaç tamamen absorbe olursa, biyolojik olarak yararlı doz, ilacın etiketinde kayıtlı olan dozdur

8
Etkin maddenin sistemik dolaşıma geçiş hızı önemlidir (Özellikle akut tedavide)
Buna astım atakları ve hiperglisemik şoklar örnek olarak gösterilebilir. Buna karşılık antihipertansif ilaçların yan etkilerini ortadan kaldırmak için, bu ilaçların daha düşük absorbsiyon hızına sahip olmaları istenir

9
Biyoyararlanıma bu kadar önem verilmesinin nedeni, ilaçların terapötik etkinliğinin önemli oranda etkin maddenin fizyolojik yararlanımına bağlı olmasındandır. Biyoeşdeğerlik yerine, klinik eşdeğerliğin gösterilmesi, ideal bir amaç olmasına rağmen, pek çok durumda terapötik eşdeğerliği kantitatif olarak saptamak çok zordur. Ayrıca birey içi ve bireyler arası değişkenlik nedeniyle klinik çalışmalarda çok sayıda deneğe ihtiyaç olup ekonomik açıdan çok pahalıdır.

10
BİYOEŞDEĞERLİK Bizim yönetmeliğimizde yok !! Aynı EM aynı miktarda içeren fakat farklı imalatçılar tarafından farklı yardımcı madde ve farklı teknolojilerle hazırlanmış ve ticari isimleri de farklı olan müstahzarlar KİMYASAL EŞDEĞER kabul edilir Algo, Asabrin, ASP, Aspinal, Aspirin, Asporan, Ataspin, Ecoprin, aynı miktarda ASA içeriyor... 2 farklı müstahzar aynı etkin maddenin veya maddelerin aynı miktarını aynı veya karşılaştırılabilir standartlara uyan farmasötik şekiller içinde içeriyorlarsa FARMASÖTİK EŞDEĞER kabul edilir

11
Yeterince yakın biyoyararlanım gösteren farmasötik eşdeğer
Benzer biyoyararlanımlı + farmasötik eşdeğer %80-125 BİYOEŞDEĞER Farmasötik eşdeğer olan iki müstahzarın, aynı molar dozda verilişinden sonra biyoyararlanımlarının (hız ve derece) ve böylece etkilerinin hem etkinlik, hem güvenlik bakımından esas olarak aynı olmasını sağlayacak derecede benzer olması Yeterince yakın biyoyararlanım gösteren farmasötik eşdeğer
 ve böylece etkilerinin hem etkinlik, hem güvenlik bakımından esas olarak aynı olmasını sağlayacak derecede benzer olması. Yeterince yakın biyoyararlanım gösteren farmasötik eşdeğer.")
12
Biyoeşdeğer preparatlar da bu tanım kapsamında değerlendirilebilir
TERAPÖTİK EŞDEĞER Bir müstahzarın, etkinliği ve güvenilirliği daha önce tespit edilmiş bir başka müstahzar ile aynı etkin maddeyi veya terapötik molekül kısmını içermesi ve aynı etkinlik ve güvenliliği klinik olarak göstermesi hali Benzer klinik yanıtı gösteren farmasötik eşdeğer ilaçlar KLİNİK EŞDEĞER kabul edilir

13
Kandaki EM çekirdeği derişimlerinin biyoeşdeğer olması kaydıyla
Müstahzardaki etkin maddenin kimyasal olarak aynı çekirdek olmakla birlikte, farklı türevler, farklı tuzlar, farklı dozaj şekilleri veya farklı dozlarda içeren ilaçlar FARMASÖTİK SEÇENEK (ALTERNATİF) kabul edilir 1- Eritromisin etil süksinat –eritromisin stearat (farklı türev) 2- Tetrasiklin HCl ile tetrasiklin fosfat (farklı tuz) 3- Parasetamol tablet ile parasetamol kapsül (farklı dozaj şekli) 4- Aspirin tb. 500mg ile Aspirin Bebek çiğneme tb. 100mg (farklı dozaj şekli-farklı doz) Kandaki EM çekirdeği derişimlerinin biyoeşdeğer olması kaydıyla
 kabul edilir. 1- Eritromisin etil süksinat –eritromisin stearat (farklı türev) 2- Tetrasiklin HCl ile tetrasiklin fosfat (farklı tuz) 3- Parasetamol tablet ile parasetamol kapsül (farklı dozaj şekli) 4- Aspirin tb. 500mg ile. Aspirin Bebek çiğneme tb. 100mg (farklı dozaj şekli-farklı doz) Kandaki EM çekirdeği derişimlerinin biyoeşdeğer olması kaydıyla.")
14
ORAL İLAÇ UYGULAMASINDA BİYOYARARLANIM

15
İLAÇ Hastalıkların tanı ve tedavisinde ayrıca hastalıklardan korunma amacı ile kullanılan madde Farmakokinetik: Organizmaya verilişten sonra (plazma, kan, idrar, tükürük, lenf ve omurilik sıvısı) biyolojik sıvılarda oluşturduğu profilleri inceler ve bu profilleri matematiksel denklemlerle tanımlar
 biyolojik sıvılarda oluşturduğu profilleri inceler ve bu profilleri matematiksel denklemlerle tanımlar.")
16
??? 1-İlaçlARIN EMİLİMİ İlk geçiş etkisi
Emilim: Uygulama bölgesinden kan dolaşımına geçiş A: ENTERAL 1- Oral 2- Sublingual 3- Rektal B: PARENTERAL 1- İntravasküler intravenöz intraarteriyel 2- İntramusküler 3- Subkütan C: DİĞER UYGULAMA YOLLARI 1- İnhalasyon 2- İntranazal 3- İntratekal/intraventriküler 4- Topik 5- Transdermal Emilim hızı ve etkinliği uygulama yoluna bağlıdır İLAÇ UYGULAMA YOLLARI Uygulama yolu ilacın-etkin maddenin çözünürlüğüne, tedavi amacına (hızlı-yavaş, uzun-kısa etki)
")
17
A: İlaçların GİS’ten taşınması 1- Pasif Difüzyon 2- Aktif Transport
İlaçlARIN EMİLİMİ A: İlaçların GİS’ten taşınması 1- Pasif Difüzyon 2- Aktif Transport B: pH’nın EM emilimine etkisi C: Emilimi etkileyen fiziksel etkenler 1- Emilim bölgesindeki kan akımı 2- Emilimin gerçekleştiği yüzeyin alanı 3- Emilim yüzeyi ile temas süresi

18
İlaçlARIN EMİLİMİ

19
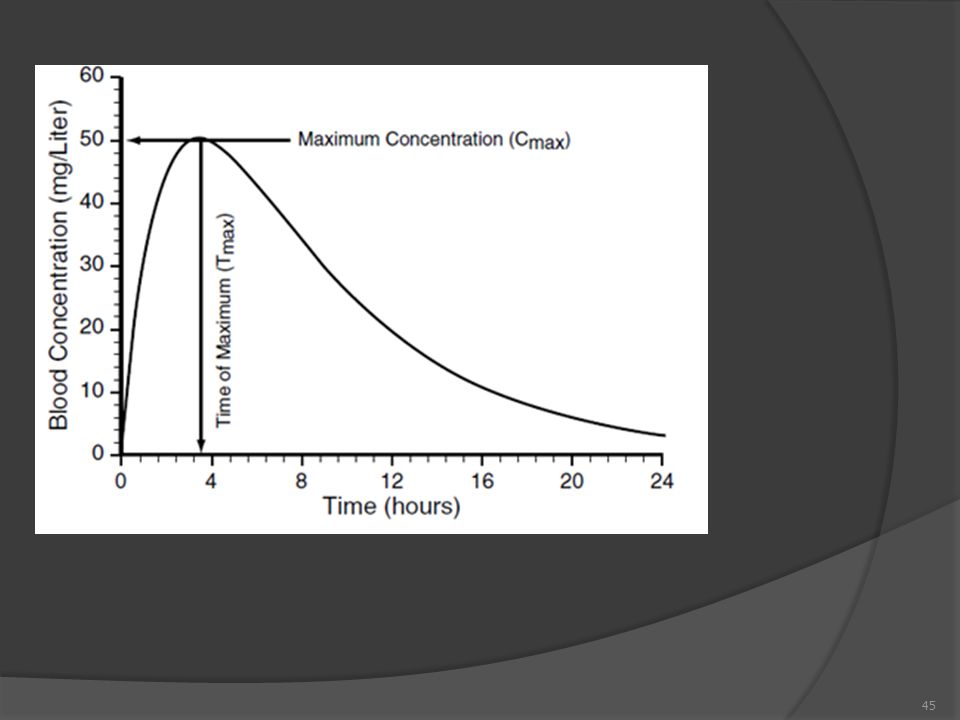
Biyoyararlanım incelemelerinde, etkin maddenin (ilacın) veya etkin metabolitlerin ve gereken durumlarda ilaç molekülünün etkin kısmının, esas olarak plazma konsantrasyonu-zaman eğrisi belirlenerek, bu eğrinin altındaki alan (EAA) en az üç yarılanma ömrüne eşit bir süre boyunca ölçülmek ve bu eğri ile ilgili olan doruk plazma konsantrasyonu (Cmax) ve doruk plazma konsantrasyonuna erişme süresi (tmax) ölçülmek suretiyle değerlendirme yapılır. Kan örneği alma zamanları ve araları ölçülecek karakteristiklerin yeterli derecede ayrıntılı bir zaman seyrini ortaya koyabilecek şekilde seçilmelidir.

20
Biyoyararlanımın belirlenmesi

21
İlave olarak veya şartlar gerektirdiği takdirde sadece kümülatif renal itrah edilen miktar (Ae) itrah hızı (dAe/dt) ve tümüyle absorbe edilip itrah edilme süresi (t) ölçülerek değerlendirme yapılabilir. Kararlı durum (plato düzeyi) incelemelerinde doz aralığı boyunca, eğri altındaki alan (EAA)= ve dalgalanma genliği hesaplanmasına gerek olup olmadığına karar verilir. Gerekli olan durumlarda ilave olarak, farmakodinamik tesirleri zamana göre ölçmek suretiyle değerlendirme yapılır. Bu takdirde ölçümler yeterli derecede ayrıntılı bir zaman seyri sağlamalı ve başlangıç değerleri aynı olmalıdır. Ölçümlerin özgüllüğü, kesinliği (prezisyonu) ve tekrarlanabilirliği yeterli derecede olmalıdır. Doz/cevap ilişkisinin non-lineer karakteri dikkate alınmalıdır
 incelemelerinde doz aralığı boyunca, eğri altındaki alan (EAA)= ve dalgalanma genliği hesaplanmasına gerek olup olmadığına karar verilir. Gerekli olan durumlarda ilave olarak, farmakodinamik tesirleri zamana göre ölçmek suretiyle değerlendirme yapılır. Bu takdirde ölçümler yeterli derecede ayrıntılı bir zaman seyri sağlamalı ve başlangıç değerleri aynı olmalıdır. Ölçümlerin özgüllüğü, kesinliği (prezisyonu) ve tekrarlanabilirliği yeterli derecede olmalıdır. Doz/cevap ilişkisinin non-lineer karakteri dikkate alınmalıdır.")
22
Kan dolaşımından hücreler arası alan ve/veya doku hücrelerine geçiş
2-İlaçlARIN DAĞILIMI Geri dönüşümlü Kan dolaşımından hücreler arası alan ve/veya doku hücrelerine geçiş İlacın plazmadan hücreler arası alana geçmesi özellikle kan akımı, kılcal damarların geçirgenliği, ilacın doku ve plazma proteinlerine bağlanma derecesi ve ilacın nispeten hidrofobik özelliğine bağlıdır

23
Farklı doku ve organlarda kan akımı değişken
2- Kılcal damarların geçirgenliği Kılcal damarların yapısı (Şekil 4-5) Endotel hücreleri arasındaki yarıklar a.1.) BBB b) İlacın yapısı Elektron dağılımı düzenli + elektrik yükü yok + hidrofobik = GEÇEBİLİR 3- İlacın doku ve plazma proteinlerine bağlanma derecesi Geri dönüşümlü bağlanma dolaşımda daha uzun süre kalmayı sağlar Bağlanma genellikle albumine olur ve depo görevi görür Karaciğer Beyin
 Endotel hücreleri arasındaki yarıklar. a.1.) BBB. b) İlacın yapısı. Elektron dağılımı düzenli + elektrik yükü yok + hidrofobik = GEÇEBİLİR. 3- İlacın doku ve plazma proteinlerine bağlanma derecesi. Geri dönüşümlü bağlanma dolaşımda daha uzun süre kalmayı sağlar. Bağlanma genellikle albumine olur ve depo görevi görür. Karaciğer. Beyin.")
24
DAĞILIM HACMİ A: Vucuttaki sıvı kompartmanları
1- Plazma :(MA ve PP’ne bağlanma yüksek) 2- Hücre Dışı (extraselüler sıvı) :(Hidrofil+MA küçük yarıklardan geçerek emilir ancak hücre içine giremez) 3- Total vücut sıvısı :(Hidrofob+MA küçük yarıklardan geçerek emilir ve hücre içine de girer) 4- Diğer : Fetüs B: Sanal dağılım hacmi (Daha sonra değinilecek) Toplam sıvı 42L /70 kg *Hücre içi 28L *Hücre dışı 14L Hücreler arası 10L Plazma 4L
 2- Hücre Dışı (extraselüler sıvı) :(Hidrofil+MA küçük yarıklardan geçerek emilir ancak hücre içine giremez) 3- Total vücut sıvısı :(Hidrofob+MA küçük yarıklardan geçerek emilir ve hücre içine de girer) 4- Diğer : Fetüs. B: Sanal dağılım hacmi. (Daha sonra değinilecek) Toplam sıvı 42L /70 kg. *Hücre içi 28L. *Hücre dışı 14L. Hücreler arası 10L. Plazma 4L.")
25
İLAÇLARIN PLAZMA PROTEİNLERİNE BAĞLANMASI
A: Albuminin bağlama kapasitesi Zayıf bağlarla – 1 albumine 1 ilaç veya 1 albumine 1’den fazla ilaç (yüksek bağlanma) Zayıf asitler ve hidrofobik ilaçlara afinitesi yüksek Hidrofobik ve nötraller bağlanmaz B: İlaçlar arasında bağlanma bölgesi için yarışma Albumine yüksek afinitesi olan ilaçlar birlikte verildiğinde bağlanma bölgesi için yarışırlar Doz x Albunine bağlanma kapasitesi = YÜKSEK ve DÜŞÜK oluşuna göre 1. Grup I İlaçlar 2. Grup II İlaçlar Bağlı ilacın ayrılmasının klinik önemi
 Zayıf asitler ve hidrofobik ilaçlara afinitesi yüksek. Hidrofobik ve nötraller bağlanmaz. B: İlaçlar arasında bağlanma bölgesi için yarışma. Albumine yüksek afinitesi olan ilaçlar birlikte verildiğinde bağlanma bölgesi için yarışırlar. Doz x Albunine bağlanma kapasitesi = YÜKSEK ve DÜŞÜK oluşuna göre. 1. Grup I İlaçlar. 2. Grup II İlaçlar. Bağlı ilacın ayrılmasının klinik önemi.")
26
Grup I İlaçlar İlacın dozu albuminin bağlama kapasitesinden düşükse (Kullanılan çoğu ilaç) 2. Grup II İlaçlar İlacın dozu albuminin bağlama kapasitesinden yüksekse (İlacın çoğu serbest) Bağlı ilacın ayrılmasının klinik önemi İlaçları gruplandırmanın önemi Tolbutamid gibi I. Grup ilaçlar ile Sulfonamid antibiyotikleri gibi II. Grup ilaçlar birarda kullanıldığında ortaya çıkar Tek başına Tolbutamid (sülfanilüre yapısında ve pankreastan insülin salgılanmasını sağlıyor)’in bağlanma oranı %95 (%5 serbest) ken hiçbir farmakolojik etki gösterememektedir. Sulfonamid verildiğinde serbest Tolbutamid %100’e hızlı bir şekilde ulaşır ve hücreler arasındaki sıvıya geçerek yeni bir denge kons. oluştururur
 Bağlı ilacın ayrılmasının klinik önemi. İlaçları gruplandırmanın önemi Tolbutamid gibi I. Grup ilaçlar ile Sulfonamid antibiyotikleri gibi II. Grup ilaçlar birarda kullanıldığında ortaya çıkar. Tek başına Tolbutamid (sülfanilüre yapısında ve pankreastan insülin salgılanmasını sağlıyor)’in bağlanma oranı %95 (%5 serbest) ken hiçbir farmakolojik etki gösterememektedir. Sulfonamid verildiğinde serbest Tolbutamid %100’e hızlı bir şekilde ulaşır ve hücreler arasındaki sıvıya geçerek yeni bir denge kons. oluştururur.")
27
İlacın bağlanma bölgesinde
Grup I İlaç : Min Doz Grup II İlaç : Max Doz Grup I ve Grup II İlaç birlikte kullanıldığında Grup II verildiğinde Grup I ayrılıyor

28
C. Bağlı ilacın ayrılmasının Vd ile ilişkisi
Etkin maddenin albuminden ayrılmasının Vd de yaratacağı etki ilacın terapötik indeksine bağlı Vd büyük serbest EM konsantrasyonunda oluşan etki önemsiz Vd küçük serbest EM konsantrasyonunda oluşan etki önemli

29
v= ilaç metabolizmasının hızı= Vmax [C] / Km +[C]
3-İlaçlARIN metabolİzasyonu karaciğer Pro-drug Organizmadaki enzimler etkin maddeyi kimyasal biyotransformasyonlara uğratır ki, buna metabolizma (anabolizma+ katabolizma) denmektedir Metabolizmanın kinetiği Birinci Derece Kinetik: İlaçların metabolik transformasyonunu enzimler katalize eder ve reaksiyonların pek çoğu Michaelis-Menten Kinetiğine uyarlar v= ilaç metabolizmasının hızı= Vmax [C] / Km +[C] [C] << Km v = Vmax [C] / Km 2. Sıfırıncı Derece Kinetik: Aspirin , etanol gibi ilaçların kullanılan dozu çok yüksektir. Bu nedenle; Km << [C] v = Vmax [C] / [C]
![v= ilaç metabolizmasının hızı= Vmax [C] / Km +[C]](http://slideplayer.biz.tr/slide/1888495/7/images/29/v%3D+ila%C3%A7+metabolizmas%C4%B1n%C4%B1n+h%C4%B1z%C4%B1%3D+Vmax+%5BC%5D+%2F+Km+%2B%5BC%5D.jpg "3-İlaçlARIN metabolİzasyonu. karaciğer. Pro-drug. Organizmadaki enzimler etkin maddeyi kimyasal biyotransformasyonlara uğratır ki, buna metabolizma (anabolizma+ katabolizma) denmektedir. Metabolizmanın kinetiği. Birinci Derece Kinetik: İlaçların metabolik transformasyonunu enzimler katalize eder ve reaksiyonların pek çoğu Michaelis-Menten Kinetiğine uyarlar. v= ilaç metabolizmasının hızı= Vmax [C] / Km +[C] [C] << Km. v = Vmax [C] / Km. 2. Sıfırıncı Derece Kinetik: Aspirin , etanol gibi ilaçların kullanılan dozu çok yüksektir. Bu nedenle; Km << [C] v = Vmax [C] / [C]")
30
İlaç Metabolizması reaksiyonları
Böbreklerin hücre zarlarını geçerek distal tübüler tarafından geri emilen lipofilik ilaçları vücuttan yeterli derecede atmaları mümkün değildir. Bu nedenle yağda çözünen ilaçlar Faz I ve II adı verilen iki ana mekanizma ile önce karaciğerde metabolize edilmelidirler 1- Faz I : lipofilik moleküller –OH veya –NH2 gibi fonksiyonel gruplar kazandırarak yada fonksiyonel polar grupları ortaya çıkararak polar moleküllere çevirirler Faz I metabolizması ilaçların etkilerini arttırabilir, azaltabilir veya değiştirmeyebilir

31
2- Faz II : Konjugasyon reaksiyonları meydana gelir
2- Faz II : Konjugasyon reaksiyonları meydana gelir. Faz I’in metaboliti yeterince polar ise böbreklerden atılabilir (istisnai durumlar sözkonusu) –OH, –NH2 veya –COOH grubu içeren ilaçlar doğrudan Faz II’ye girebilirler. Yüksek derecede polar hale gelen ilaç konjugatları böbrekler veya safra yolu ile atılabilirler Bütün ilaçlar Faz I ve Faz II reaksiyonlarını sıralı geçirmezler. Faz II ile başlayıp sonradan Faz I’e uğrayabilir
 –OH, –NH2 veya –COOH grubu içeren ilaçlar doğrudan Faz II’ye girebilirler. Yüksek derecede polar hale gelen ilaç konjugatları böbrekler veya safra yolu ile atılabilirler. Bütün ilaçlar Faz I ve Faz II reaksiyonlarını sıralı geçirmezler. Faz II ile başlayıp sonradan Faz I’e uğrayabilir.")
32
4-İlaçlARIN elİmİnasyonu
etkin madde vücuttan idrar, ter, tükürük, akciğerler, safra-feçes gibi atılım yoluyla uzaklaştırılır İlaçların böbrekler yoluyla atılımı 1. Glomerüler Filtrasyon 2. Proksimal tübüler sekresyon 3. Distal tübüler geri emilim 4. İlaç metabolizmasının rolü

33
BİYOYARARLANIM ÖLÇÜTLERİ
1- Eğri Alında Kalan Alan (EAA, AUC) 2- Plazma Doruk Konsantrasyonu (Cmax) 3- Plazma Doruk süresi (tmax) 4- İdrarla atılan toplam EM Miktarı (Au∞) 5- İdrarla Atılma Hızları (ΔAu/Δt)
 2- Plazma Doruk Konsantrasyonu (Cmax) 3- Plazma Doruk süresi (tmax) 4- İdrarla atılan toplam EM Miktarı (Au∞) 5- İdrarla Atılma Hızları (ΔAu/Δt)")
34
1- Eğri Alında Kalan Alan (EAA, AUC)
11- Eğri altında kalan alan (AUC, E AA) Plazma derişim-zaman eğrisinin (plazma profilinin) altında kalan alandır. İki tane alan sözkonusudur: 1) Sıfırdan sonsuza alan (AUC ): Eğri altındaki tüm alandır 0 ∞ 2) Son deneysel noktaya kadar olan alan (AUC^j; 0 n last Son deneysel noktaya kadar olan alandır. Güvenilir olabilmesi için değeri, AUC'nin en az % 80'i olmalıdır. 2) Son deneysel noktaya kadar olan alan (AUC):
 Plazma derişim-zaman eğrisinin (plazma profilinin) altında kalan alandır. İki tane alan sözkonusudur: 1) Sıfırdan sonsuza alan (AUC ): Eğri altındaki tüm alandır. 0 ∞ 2) Son deneysel noktaya kadar olan alan (AUC^j; 0 n last. Son deneysel noktaya kadar olan alandır. Güvenilir olabilmesi için değeri, AUC nin en az % 80 i olmalıdır. 2) Son deneysel noktaya kadar olan alan (AUC):")
35
AUC hesaplama yöntemleri
1- İntegral Yöntemi 2- Trapez Yöntemi (yamuk yöntemi) Doğrusal AUC b) Logaritmik AUC
 Doğrusal AUC. b) Logaritmik AUC.")
36
1- İntegral Yöntemi

37
Plazma derişimi-zaman eğrisi
AUC

38
Assessing Bioavailability of Drug Delivery Systems, 2005
Oral AUC hesaplaması Assessing Bioavailability of Drug Delivery Systems, 2005

39
2- Trapez Yöntemi (yamuk yöntemi) Doğrusal Trapez Yöntemi
Logaritmik Trapez Yöntemi Doğrusal trapez yönteminde AUC hesaplanırken veri noktaları doğrusal olarak enterpole edilir. FDA ve OGD (Other Government Departments) tarafından kabul edilen bir hesaplama yöntemi
 tarafından kabul edilen bir hesaplama yöntemi.")
40
b) Logaritmik Trapez Yöntemi
Logaritmik trapez yönteminde AUC hesaplanırken veri noktaları logaritmik olarak enterpole edilir. Daha kesin sonuçlar elde edilir Yöntemlerin birlikte kullanıldığı ve ayrıca farklı bir yöntem olan Simpson Yöntemi de AUC hesaplanırken kullanılabilir Simpson Yöntemi

41
ÖRNEK

42
Emilim hız değişmezi (ka)
İlaç şekilleri organizmaya emilmeli yollardan verildiğinde, emilim hız değişmezi ortaya çıkacaktır. İlaçlar büyük oranda oral yoldan verildiklerinden, bu parametre oral emilim hız değişmezi olarak rol oynayacaktır. Diğer emilmeli yolların da (rektal, transdermal, vajinal, İ.M., nazal, v.b.) kendilerine özgü emilim hız değişmezleri olması söz konusudur
 kendilerine özgü emilim hız değişmezleri olması söz konusudur.")
43
Denetimli salım preparatı
Zincirleme kompartman modeli Denetimli salım preparatı kr0 veya kr1 GİS ka Kan kd = km + kel Yükleme Dozu Uzaklaşma ka« kr1 veya kr Kinetik olayın şeffaflaşması

44
2- Plazma Doruk Konsantrasyonu (Cmax)
Plazmada izlenen en yüksek derişimdir. Oldukça önemli bir parametredir. Emilim hızının bir göstergesi olarak kabul edilir. Ayrıca biyoyararlanımda da değerlendirilir. İki tane Cmax söz konusudur: Deneysel Cmax : Deneysel verilerden elde edilen en yüksek değerdir (Resmi otoriteler bunu kabul ederler. Ancak bu, farmakokinetik deneyin iyi tasarlanmış olmasını gerektirir. Eğer deneysel noktalar arasında çok aralıklar varsa, gerçek Cmax iki nokta arasında kalabilir)
")
46
2) Kestirim Cmax: Farmakokinetik modelleme sonrasında elde edilen denklemden bulunan Cmax 'tır. Dikkatli şekilde yapılırsa, daha tutarlı bir sonuç vermesi beklenir. Cmax, doğal olarak alınan dozun bir fonksiyonu olup, doğrusal farmakokinetiğe uyan ilaçlar için doza paralel olarak değişir. Dolayısıyla doza bağımlı bir farmakokinetik parametredir

47
Assessing Bioavailability of Drug Delivery Systems, 2005
3- Plazma Doruk süresi (tmax) Plazma seviyesinin Cmax'a çıkabilmesi için gerekli olan süre kalitatif bir biyoyararlanım parametresidir. Bunun nedeni, tmax'ın kolay saptanamamasıdır Değeri, çoğu oral ilaç için 2-4 saat dolaylarında olmakla beraber, deneklerarası varyansı yüksektir İlaçla beraber alınan besin miktarı ve türü bu değeri etkilemektedir Emilme hızıyla ters orantılı bir parametredir Assessing Bioavailability of Drug Delivery Systems, 2005
 Plazma seviyesinin Cmax a çıkabilmesi için gerekli olan süre. kalitatif bir biyoyararlanım parametresidir. Bunun nedeni, tmax ın kolay saptanamamasıdır. Değeri, çoğu oral ilaç için 2-4 saat dolaylarında olmakla beraber, deneklerarası varyansı yüksektir. İlaçla beraber alınan besin miktarı ve türü bu değeri etkilemektedir. Emilme hızıyla ters orantılı bir parametredir. Assessing Bioavailability of Drug Delivery Systems,")
48
4- İdrarla atılan toplam EM Miktarı (Au∞)
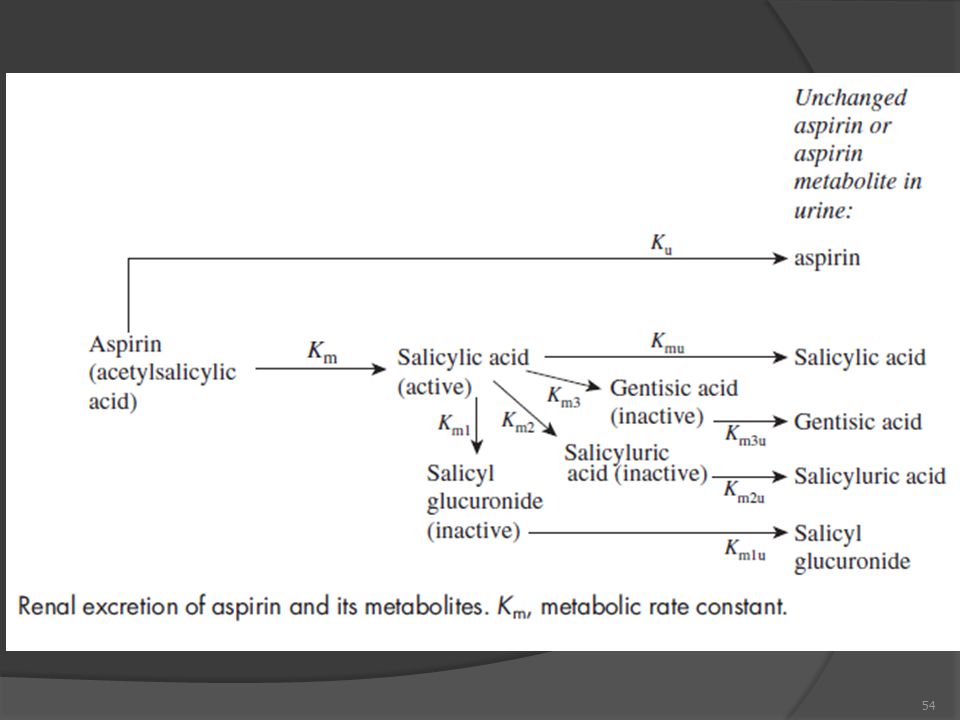
5- İdrarla Atılma Hızları (ΔAu/Δt) a- Metabolizma Hız Değişmezi (km) Metabolizma yolaklarının reaksiyonları paralel reaksiyonlar hız değişmezleri aritmetik olarak toplanır ve metabolizma genel hız değişmezini oluşturur: km = km1+km2+...+kmn [Burada km, metabolizma reaksiyonlarının tümünü tanımlamaktadır] b- Atılım Hız Değişmezi (kel) Değişik yolların her birinin hız değişmezleri paralel reaksiyonlar olması dolayısıyla toplanmaktadır kel = kel1+kel2+...+keln Gerek metabolizma ve gerekse atılım olayları da paraleldir. Dolayısıyla hız değişmezleri toplanarak uzaklaşma olayını oluştururlar.
 a- Metabolizma Hız Değişmezi (km) Metabolizma yolaklarının reaksiyonları paralel reaksiyonlar. hız değişmezleri aritmetik olarak toplanır ve metabolizma genel hız değişmezini oluşturur: km = km1+km2+...+kmn [Burada km, metabolizma reaksiyonlarının tümünü tanımlamaktadır] b- Atılım Hız Değişmezi (kel) Değişik yolların her birinin hız değişmezleri paralel reaksiyonlar olması dolayısıyla toplanmaktadır. kel = kel1+kel2+...+keln. Gerek metabolizma ve gerekse atılım olayları da paraleldir. Dolayısıyla hız değişmezleri toplanarak uzaklaşma olayını oluştururlar.")
49
c- Uzaklaşma Hız Değişmezi (kd, β veya λ1)
Genel uzaklaşma hız değişmezi, olmaktadır. kd = km + kel Burada kd, etkin maddenin plazmadan genel çıkışını, yani bütün atılım ve metabolizma yollarının tümünü kapsamaktadır. Birimi, zaman-1’ dir (genellikle saat-1 olarak kullanılır). Emilim ve dağılım bittikten sonra sadece uzaklaşma emilim ve uzaklaşma olayları beraber t* Emilimin bittiği kabul edilir
. Emilim ve dağılım bittikten sonra. sadece uzaklaşma. emilim ve uzaklaşma olayları beraber. t* Emilimin bittiği kabul edilir.")
50
Assessing Bioavailability of Drug Delivery Systems, 2005
d- Biyolojik Yarı Ömür (t1/2) Etkin maddenin yarısının organizmayı 'terk etmesi' için gereken süre t1/2= ln2 / kd = 0.693/ kd Etkin maddelerin biyolojik yarı-ömürleri birkaç dakikadan 10 güne kadar değişmektedir (İstisnalar hariç) Assessing Bioavailability of Drug Delivery Systems, 2005
 Etkin maddenin yarısının organizmayı terk etmesi için gereken süre. t1/2= ln2 / kd = 0.693/ kd. Etkin maddelerin biyolojik yarı-ömürleri birkaç dakikadan 10 güne kadar değişmektedir (İstisnalar hariç) Assessing Bioavailability of Drug Delivery Systems,")
51
e- Ortalama Yaşam Süresi (Mean Residence Time ;MRT)
Organizmaya verilmiş etkin madde moleküllerinin her biri organizmayı değişik zamanlarda terk eder. Bütün moleküllerin organizmada kaldıkları sürelerin ortalaması alınabilseydi, bu parametre ortaya çıkardı MRT iv = 1/ kd MRT iv = t1/2 / ln2 = t1/2 / 0.693

52
Vd ext = D / Co Vp = D / Co f- Sanal Dağılım Hacmi (Vd)
Tek kompartımanlı model için bütün vücut homojen bir kompartıman gibi düşünülmektedir Etkin maddeler organizmaya verildiklerinde bütün vücuda dağılırlar ve plazmada da belli bir derişim oluştururlar Şekil 11 Plazma haricindeki bölümlere de dağıldığı için çok yüksek değerler alabilir = sanal dağılım hacmi Vd ext = D / Co Yalnızca plazmada dağılıyorsa Vp = D / Co Şekil 12

53
Cl = kd Vd g- Klirens (Cl, arınma)
Organizmadaki sıvıların, birim zamanda etkin maddeden arınmış hacmi (mL.dk-1, L.saat-1) Uzaklaşma hızı (miktar.zaman-1) = dA/dt = CI*C Cl= DİV / AUC 0 ∞ Cl = kd Vd
 Uzaklaşma hızı (miktar.zaman-1) = dA/dt = CI*C. Cl= DİV / AUC. 0 ∞ Cl = kd Vd.")
55
kd = Cl / Vd Hepatik ve renal klirensin toplamı
the THERAPEUTIC ıNDEX is the ratio relating the median lethal dose (the concentration causing the deaths of 50% of experimental animals) and the median effective dose (the concentration at which the drug is effective in 50% of cases): kd = Cl / Vd [Cl, klirens ve Vd, sanal dağılım hacmi] Assessing Bioavailability of Drug Delivery Systems, 2005
 and the median effective dose (the concentration at which the drug is effective in 50% of cases): kd = Cl / Vd. [Cl, klirens ve Vd, sanal dağılım hacmi] Assessing Bioavailability of Drug Delivery Systems,")
56
Kinetics of the drug in various compartments
1. Amount of drug along the GIT 2. Amount of drug in the blood 3. Amount of drug eliminated

57
BİYOYARARLANIM BİYEŞDEĞERLİK ÇALIŞMALARININ PLANLANMASI

58
Biyoyararlanım çalışmasının planlanması
Araştırmacı Klinik sorumlu Analizci Biyoistatistikci İzleyici GCP ve GLP Yerel Etik Kurul Ana Etik Kurul Biyoyararlanım çalışmasının planlanması

59
Biyoyararlanım çalışması;
Dar terapötik index’e Doza bağlı ciddi toksisiteye Non lineer farmakokinetiğe sahip ilaçlar için gereklidir Aşırı Biyoyararlanım (Suprabioavailability) : Yeni ilacın BY onaylanmış ilaçtan yüksek ise Daha düşük dozlu formülasyonun hazırlanması gerekmektedir i.v. uygulanacak basit çözeltiler İnhale edilecek gazlar Sistemik etki amaçlanmamış topik uygulamalar (oküler, vajinal, dermal, nazal vs) sahip ilaçlar için BY çalışmasına gerek yoktur
 : Yeni ilacın BY onaylanmış ilaçtan yüksek ise. Daha düşük dozlu formülasyonun hazırlanması gerekmektedir. i.v. uygulanacak basit çözeltiler. İnhale edilecek gazlar. Sistemik etki amaçlanmamış topik uygulamalar (oküler, vajinal, dermal, nazal vs) sahip ilaçlar için BY çalışmasına gerek yoktur.")
60
Kadın denekler hamile olmamalı Çocuk denekler Yaşlı denekler Hastalar
Deneklerin Seçimi Standardizasyon Sağlıklı Gönüllüler Kadın denekler hamile olmamalı Çocuk denekler Yaşlı denekler Hastalar Hareket Gıda Sigara alışkanlığı Alkol kullanımı Başka ilaç kullanımı Acil durumlar İstatistiksel yorum Denek Ölçütleri Yaş (18-55) Sağlık durumu Boy/Ağırlık oranı (normalin %15’i) 10h açlık gerekli Uygulamayı takiben 4h yemek yok İlacın yemekle alınması önerildi ise Kcal %50’si yağ’dan %15-20 protein Kalan miktar karbonhidrat Denek Sayısı Min 12 Sorunlu ilaç 16(18)-24 + yedek 1-2 Her denekten ortalama örnek alınır Drop-out Denek ilavesi : en az 12 denek Anlamlılık düzeyi
 Sağlık durumu. Boy/Ağırlık oranı (normalin %15’i) 10h açlık gerekli. Uygulamayı takiben 4h yemek yok. İlacın yemekle alınması önerildi ise Kcal. %50’si yağ’dan. %15-20 protein. Kalan miktar karbonhidrat. Denek Sayısı. Min 12. Sorunlu ilaç 16(18) yedek 1-2. Her denekten ortalama örnek alınır. Drop-out. Denek ilavesi : en az 12 denek. Anlamlılık düzeyi")
61
Deneklerin genotip veya fenotipleri dikkate alınmalıdır
Yaşlı deneklerde kullanılacak ilaç için 60 ve daha yaşlı Kadın erkek eşit oranda Toksisite riski olan ilaçlar durumları stabil olan hasta denekler üzerinde denenebilir Zararlı yan etkileri olan ilaçlarda denekler sınırlandırılabilir Örn: teratojenik bir ilaç yalnızca erkekler üzerinde denenebilir Özellikle erkeklere veya özellikle kadınlara yönelik hazırlanan ilacın gerektirdiği denek gurubu çalışmada belirtilmelidir Deneklerin genotip veya fenotipleri dikkate alınmalıdır

62
Örnekleme zamanları genelde t1/2‘den uzun olmamalıdır
Deneyin yürütülmesi Kan- idrar min (3 x t1/2) izlenmeli Min 10 veri noktası Emilim aşamasında yeter sayıda nokta (min 3-4) Cmax yeter sayıda nokta (min 3-4) Atılım aşaması nokta (Noktalar belirlenirken AUC, Cmax ve kd dikkate alınmalı) AUCo-n AUC o-∞ %80 den az olmamalı Wash-out süresi x t1/2 Çapraz çalışmalar haftanın aynı gün ve saatinde İdrar örneği yeterince sık (1-2 h aralıkla) Değişmemiş ilaç veya metabolitler izlenir Örnekleme zamanları genelde t1/2‘den uzun olmamalıdır
 izlenmeli. Min 10 veri noktası. Emilim aşamasında. yeter sayıda nokta (min 3-4) Cmax yeter sayıda nokta (min 3-4) Atılım aşaması 5-6 nokta. (Noktalar belirlenirken AUC, Cmax ve kd dikkate alınmalı) AUCo-n AUC o-∞ %80 den az olmamalı. Wash-out süresi 5-10 x t1/2. Çapraz çalışmalar haftanın aynı gün ve saatinde. İdrar örneği yeterince sık (1-2 h aralıkla) Değişmemiş ilaç veya metabolitler izlenir. Örnekleme zamanları genelde t1/2‘den uzun olmamalıdır.")
63
Nerede analiz edilecek?
Biyoanalitik Yöntemin Geliştirilmesi Çalışma Öncesi Faz Neler analiz edilecek? Etkin madde Metabolitler Pro-drug için metabolit Nerede analiz edilecek? Kan, Plazma, Serum, İdrar, Diğer biyolojik sıvılar (tükürük, BOS vs) İlaç veya metabolitlerin biyoanalitik sıvıdaki kararlılığı Analitik yöntem kararlılığı Validasyon Seçicilik (Specificity) Doğruluk (Accuracy) (± %15) Kesinlik(%15-20) Saptama sınırı (LOD) (<%20) Duyarlılık (LOQ) (<%20) Geri kazanım (ekstraksiyon) Dizi ve Doğrusallık (Range and Linearity) Çalışma Fazı İlaç veya metabolitlerin biyoanalitik sıvıdaki kararlılığı (deney sırasındaki)- saklama şartları-dondurma 3 kez Çalışmada standart eğri oluşturularak bu eğri altında kalan aralıkta örneklerin oluşturulması gerekir Sonuçlar uygun istatistiksel yöntem ile değerlendirilmelidir (ANOVA, Student-t Test vs.) Genellikle %90 güven aralığı
 İlaç veya metabolitlerin biyoanalitik sıvıdaki kararlılığı. Analitik yöntem kararlılığı. Validasyon. Seçicilik (Specificity) Doğruluk (Accuracy) (± %15) Kesinlik(%15-20) Saptama sınırı (LOD) (<%20) Duyarlılık (LOQ) (<%20) Geri kazanım (ekstraksiyon) Dizi ve Doğrusallık (Range and Linearity) Çalışma Fazı. İlaç veya metabolitlerin biyoanalitik sıvıdaki kararlılığı (deney sırasındaki)- saklama şartları-dondurma 3 kez. Çalışmada standart eğri oluşturularak bu eğri altında kalan aralıkta örneklerin oluşturulması gerekir. Sonuçlar uygun istatistiksel yöntem ile değerlendirilmelidir. (ANOVA, Student-t Test vs.) Genellikle %90 güven aralığı.")
64
Farmakokinetik parametrelerin kabul aralığı
AUC-oranı : %90 güven aralığı kabul -aralığında olmalıdır. Terapötik indeksin dar olduğu özel durumlarda kabul aralığı daraltılabilir. Cmax-oranı : %90 güven aralığı kabul aralığında olmalıdır. Terapötik indeksin dar olduğu özel durumlarda kabul aralığı daraltılabilir. Belirli durumlarda daha geniş bir aralık kabul edilebilir (örn ). [Terapötik indeksi dar olan ilaçlar için EMEA (2001) hem AUC hem Cmax için kabul aralığının daraltılabileceğini belirtirken FDA bu ilaçlar için de aralığının kullanılmasını önermektedir]
. [Terapötik indeksi dar olan ilaçlar için EMEA (2001) hem AUC hem Cmax için kabul aralığının daraltılabileceğini belirtirken FDA bu ilaçlar için de aralığının kullanılmasını önermektedir]")
65
Biyoyararlanım/Biyoeşdeğerlik Çalışmasının Planlanması
1- Referans Seçimi Piyasadaki innovatör ürün Kendini kanıtlamış ürün (BY çalışması olan) Sulu çözelti (i.v. Çözelti) Doz (eşdeğer molar doz) [sorunsuz ilaçlar için tek doz- sorunlu ilaçlar için her doz seviyesi] Yakın büyüklükteki seriler (pilot değil imalat serisi olmalı) 2- Çalışma tasarımı Çapraz çalışma (Cross over design) (aynı denekler wash out sonrası diğer ilacı kull.) Rastgele tasarım (Randomized design) (ilk 6 denek A ilacını- diğer 6 denek B ilacını kullanıyor sonra 15 gün sonra ilk 6 denek B ilacını- diğer 6 denek A ilacını kullanıyor ) Dengeli tamamlanmamış blok tasarım (Balanced incomplete block design)
 Sulu çözelti (i.v. Çözelti) Doz (eşdeğer molar doz) [sorunsuz ilaçlar için tek doz- sorunlu ilaçlar için her doz seviyesi] Yakın büyüklükteki seriler (pilot değil imalat serisi olmalı) 2- Çalışma tasarımı. Çapraz çalışma (Cross over design) (aynı denekler wash out sonrası diğer ilacı kull.) Rastgele tasarım (Randomized design) (ilk 6 denek A ilacını- diğer 6 denek B ilacını kullanıyor sonra 15 gün sonra ilk 6 denek B ilacını- diğer 6 denek A ilacını kullanıyor ) Dengeli tamamlanmamış blok tasarım (Balanced incomplete block design)")
66
3- Kör Çalışma 4- İdrar Çalışması Rastgeleleştirme Deneğin bilmemesi
Denekler rastgele sıralanır deneklere kod, sıra ve tarih verilmeli Bütün denekler raporda yer alır (ayrılanlar dahil) Deneğin bilmemesi Denetimcinin bilmemesi Analizcinin bilmemesi Çalışanların bilmemesi Çok dozlu çalışma; Emilim hızı farklı emilim oranı aynı ise Tek doz kan örnekleri analiz yöntemine göre çok düşükse (BQL) Denetimli salım prep. Non lineer kinetiğe sahip ise gereklidir 4- İdrar Çalışması Kan düzeyleri çok düşükse İlacın %40’tan fazlası değişmeden atılıyorsa idrar çalışması yapılabilir
 Deneğin bilmemesi. Denetimcinin bilmemesi. Analizcinin bilmemesi. Çalışanların bilmemesi. Çok dozlu çalışma; Emilim hızı farklı emilim oranı aynı ise. Tek doz kan örnekleri analiz yöntemine göre çok düşükse (BQL) Denetimli salım prep. Non lineer kinetiğe sahip ise gereklidir. 4- İdrar Çalışması. Kan düzeyleri çok düşükse. İlacın %40’tan fazlası değişmeden atılıyorsa idrar çalışması yapılabilir.")
67
BİYOYARARLANIM VE İLAÇ KAN DÜZEYİNDEKİ DEĞİŞİKLİKLER
Pek çok ilaç için farmakolojik yanıt ve terapötik etkinlik doğrudan ilacın gözlenen kan konsantrasyonuyla ilişkilidir Bir ilacın biyoyararlanımı, öncelikle ilacın veriliş yolu ve dozaj şekline bağlıdır İlaçların en önemli veriliş yolu oral yol olduğu için özellikle oral yolla kullanılan dozaj şekillerinin BY’lanımlarınden bahsedilmektedir İlacın kana geçiş hız ve derecesi birbirini takip eden hız proseslerine bağımlıdır Bu proseslerden en yavaşı, hız sınırlayıcı basamak olup ilacın kana geçiş hız ve derecesini tayin eder. Hız sınırlayıcı basamak bir ilaçtan diğerine değişir

68
Biyoyararlanım faktörlerine bağlı ilaç kan düzeyi değişiklikleri
Suda çözünürlüğü zayıf olan bir ilaç için hız sınırlayıcı basamak, ilacın Gİ sıvılardaki çözünmesine bağlıdır Suda çözünen bir ilacın çözünme hızı yüksek olup, bu tip bir ilaç için hız sınırlayıcı basamak ise ilacın Gİ membrandan geçiş hızıdır Biyoyararlanım faktörlerine bağlı ilaç kan düzeyi değişiklikleri İlaca ve ilaç içeren dozaj şekline bağlı faktörler Hastayla ilişkili faktörler Absorpsiyonu etkileyen fîzyolojik faktörler Absorpsiyonu etkileyen diğer faktörler Biyoyararlanımı etkileyen faktörler 1- Karaciğerden ilk geçiş eliminasyonu 2- İlacın çözünürlüğü 3- Kimyasal kararsızlık 4- İlaç formülasyonunun yapısı

69
Biyofarmasötik sınıflandırma sistemi (FDA)
Çözünürlük Permeabilite I Yüksek II Düşük III IV

70
Çözünürlük: EM terapötik dozunun 37°C'de pH 1-8 (veya pH 1-7
Çözünürlük: EM terapötik dozunun 37°C'de pH 1-8 (veya pH 1-7.5) aralığındaki üç tamponun (tercihen pH 1.0, 4.6, 6.8) herbirinin 250 mL'sinde çözünmesi demektir. EM yüksek çözünürlüğe sahip ise biyoeşdeğerlik çalışmalarından genellikle muaf olur. Ancak polimorfizm ve partikül büyüklüğü gibi faktörler çözünme hızını etkileyeceği için bu gibi durumlarda özel dikkat gösterilmesi gerekmektedir. Permeabilite: Bir ilacın dahil olduğu permeabilite sınıfı, kütle-denge çalışmaları, mutlak biyoyararlanım çalışmaları veya intestinal perfuzyon çalışmaları yapılarak tayin edilebilir. Absorpsiyon derecesinin %90'dan fazla olması durumunda etkin maddenin permeabilitesinin yüksek olduğu kabul edilir. Yüksek permeabilitenin bir göstergesi olarak kabul edilen doğrusal ve tam absorpsiyon durumlarında biyoyararlanımın etkilenme olasılığı az ilacın permeabilite değerinin 2'nin üzerinde olması durumunda tam bir absorpsiyon beklenebilir
 aralığındaki üç tamponun (tercihen pH 1.0, 4.6, 6.8) herbirinin 250 mL sinde çözünmesi demektir. EM yüksek çözünürlüğe sahip ise biyoeşdeğerlik çalışmalarından genellikle muaf olur. Ancak polimorfizm ve partikül büyüklüğü gibi faktörler çözünme hızını etkileyeceği için bu gibi durumlarda özel dikkat gösterilmesi gerekmektedir. Permeabilite: Bir ilacın dahil olduğu permeabilite sınıfı, kütle-denge çalışmaları, mutlak biyoyararlanım çalışmaları veya intestinal perfuzyon çalışmaları yapılarak tayin edilebilir. Absorpsiyon derecesinin %90 dan fazla olması durumunda etkin maddenin permeabilitesinin yüksek olduğu kabul edilir. Yüksek permeabilitenin bir göstergesi olarak kabul edilen doğrusal ve tam absorpsiyon durumlarında biyoyararlanımın etkilenme olasılığı az. ilacın permeabilite değerinin 2 nin üzerinde olması durumunda tam bir absorpsiyon beklenebilir.")
71
Biyoeşdeğerlik incelemesi istenmeyen durumlar
Aşağıdaki şartların geçerli olması kaydıyla müstahzarın sadece etkin madde dozu bakımından fark göstermesi Terapötik doz aralığı boyunca farmakokinetiğin doğrusal olması Nitel kompozisyonun aynı olması Etkin madde ile yardımcı maddeler arasındaki oranın aynı olması veya etkin maddeyi düşük konsantrasyonlarda içeren preparatlarda (%5'ten daha az) yardımcı maddeler arasındaki oranın aynı olması Farmasötik ürünlerin aynı üretici tarafından aynı yöntemle üretilmesi Orijinal müstahzar ile biyoyararlanım ve biyoeşdeğerlik incelemesinin yapılmış olması Aynı koşullar altında çözünme hızı profillerinin benzer olması
 yardımcı maddeler arasındaki oranın aynı olması. Farmasötik ürünlerin aynı üretici tarafından aynı yöntemle üretilmesi. Orijinal müstahzar ile biyoyararlanım ve biyoeşdeğerlik incelemesinin yapılmış olması. Aynı koşullar altında çözünme hızı profillerinin benzer olması.")
72
Müstahzarın küçük değişikliklerle yeniden formüle edilmesi veya aynı üretici tarafından üretim yönteminde biyoyararlanımı etkilemeyecek bilimsel gerekçelere dayalı küçük değişiklik yapılmış olması durumudur. Biyoyararlanımın incelendiği ve in vivo ile in vitro çalışmalar arasında kabul edilebilir bir korelasyonun sağlandığı durumlarda, aynı test koşulları altında, yeni ürünün çözünme hızı onaylanmış ürünün çözünme hızma benzerlik gösterirse,

73
Jenerik ilacın halen izin verilmiş olan innovatör ürün ile aynı etkin maddeyi aynı konsantrasyonda içeren sulu bir intravenöz çözelti halinde verilecekse, diğer parenteral yollarda (intramusküler, subkütan) eğer jenerik ürün halen izin verilmiş olan innovatör ürünle aynı çözelti tipinde ise (sulu veya yağlı), aynı etkin maddenin aynı konsantrasyonunu veya karşılaştırılabilir yardımcı maddeleri içeriyorsa, Kullanılan yardımcı maddeler GİS geçişi, absorpsiyonu veya etkin maddenin in vivo stabilitesini etkilemediği müddetçe, ürünün oral çözelti halinde ve etkin maddeyi ruhsatlandırılmış oral çözelti tipi ürünle aynı konsantrasyonda içeriyorsa,

74
Ürünler lokal uygulamalarla kullanılacak ürünler olarak formüle edilmişler ise (oral, nazal, okiiler, dermal, rektal, vajinal gibi. İstenmeyen derecede kısmi absorpsiyon olasılığı varsa güvenilirlik incelemeleri istenebilir), İn vivo ve in vitro çözünme hızı arasında kabul edilebilir bir korelasyonun kurulmuş olması ve yeni ürünün in vitro çözünme hızının daha önce onaylanmış önceki ürününki ile benzer olması söz konusu ise, Müstahzar inhalasyonla uygulanacak gaz halde bir ürün ise.

75
FARMAKODİNAMİK ÇALIŞMALAR
Oluşan cevap farmakolojik veya terapötik olmalıdır Valide edilmiş yöntem kullanılmalıdır Referans veya test ürünün hiçbiri max cevap oluşturmamalıdır Oluşan cevap çift-kör çalışmada kantitatif olarak belirlenebilmeli Placebo etkisi görüldüğünde ilk kıyaslama placebo ile yapılmalıdır Çapraz çalıma yapılmalıdır Patolojik ve histolojik durumlar dikkate alınmalı İstatistiksel değerlendirme

76
İN VİTRO ÇÖZÜNME HIZI Genellikle 37ºC± 0.5ºC pH 1-8 Belirlenmiş zaman aralıkları Farmakopede bulunan yöntemler veya custom












 ÇÖZÜMLEMESİ>")








