Sunuyu indir
1
KLİNİK GENETİK’E GİRİŞ
Yrd.Doç.Dr.Özgür ALDEMİR MKU Tıp Fakültesi Tıbbi Genetik AD.

2
Multifaktöriyal Kalıtım
Genetik ve çevresel faktörlerin birlikte neden olduğu genetik malformasyonların kalıtımına denir. Yarık-dudak damak, NTD, maternal FKU, amniyotik bant sendromu, şizofreni, Tip 2 Diabet…vb. Ayrıca izole Konjenital kalp hastalıkları multifaktöriyal kalıtıma iyi bir örnektir.

3
Multifaktöriyal Kalıtılan Hastalıkların Sıklığı

4
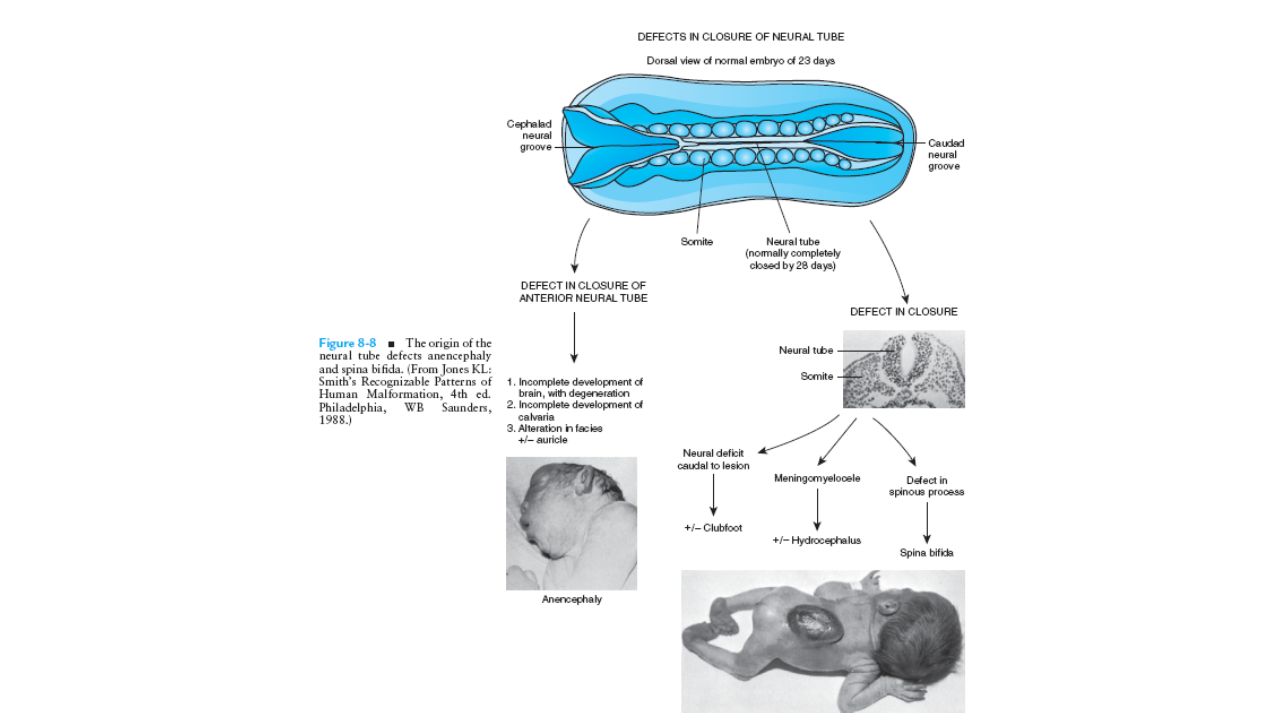
Nöral Tüp Defekti(NTD)
Genetik+Çevrenin birlikte rol oynadığı etyolojisinde maternal folat eksikliğinin suçlandığı multifaktöriyal bir hastalıktır. Spina bifida occulta, anensefaliye kadar giden geniş bir yelpazede fenotip görülebilir. İzole vakalar daha sık görülür, ek anomali varlığında sendromik olduğu akla gelmelidir. Nöral tüp defektleri şiddetine göre her gebelikte tekrarlama riski yüksektir. Prenatal dönemde maternal AFP yüksekliği ve fetal ultrason ile NTD tanısı konur.

6
Frajil X sendromu Frajil X sendromu, mental retardasyonun en sık kalıtsal sebebidir, makro testis, otistik davranış modelleri ve hiperaktivite, el ısırma, göz teması kuramama, uzun yüz ve büyük kulaklar izlenir. FMR1 geni mutasyonlarının çoğu 5’ucunda CGG tekrar sayısı artmasıyladır. Tam mutasyon tekrar s. 230 dan fazladır. Premutasyon CGG tekrarıdır. Premutasyonlu bir anne çocuğunun olması riski premutasyon büyüklüğü, fetusun cinsi ve aile hikayesine bağlıdır. Frajil X sendromu tedavisi eğitim,davranış problemlerinin farmakolojik olarak düzeltilmesiyledir.

7
CHARGE SENDROMU CHARGE-Coloboma of iris, Heart defect, Atrezi of Choana, Retardation of growth, Genital anomalies, Ear anomality. OD kalıtılan CHD7 gen mutasyonu sonucu oluşan ve 1/ sıklıkta görülür. Hasta bireyin sonraki kuşaklarda hastalığın görülme oranı %50’dir. Dış kulak anomalisi tipiktir.

8
Holoprozensefali Holoprozensefali(HPE), tek santral kesici diş ve beyin dokusunun orta hat defektleri ile giden bir durumdur. Fenotipi hafiften ağıra doğru belirgin farklılık gösterir Nonsendromik ailesel olgular 3 grubta sınıflanır. Semilobar(%28), Lobar(%9) ve alobar HPE (%63) SHH geninde mutasyon sonucu oluşur. Ayrıca ZIC2, SIX3,TGIF genleri suçlanmaktadır.
, Lobar(%9) ve alobar HPE (%63) SHH geninde mutasyon sonucu oluşur. Ayrıca ZIC2, SIX3,TGIF genleri suçlanmaktadır.")
9
Farklı Toplumlardaki Otozomal Hastalıkların insidansı,Gen sıklığı ve Heterozigotların Oranı

10
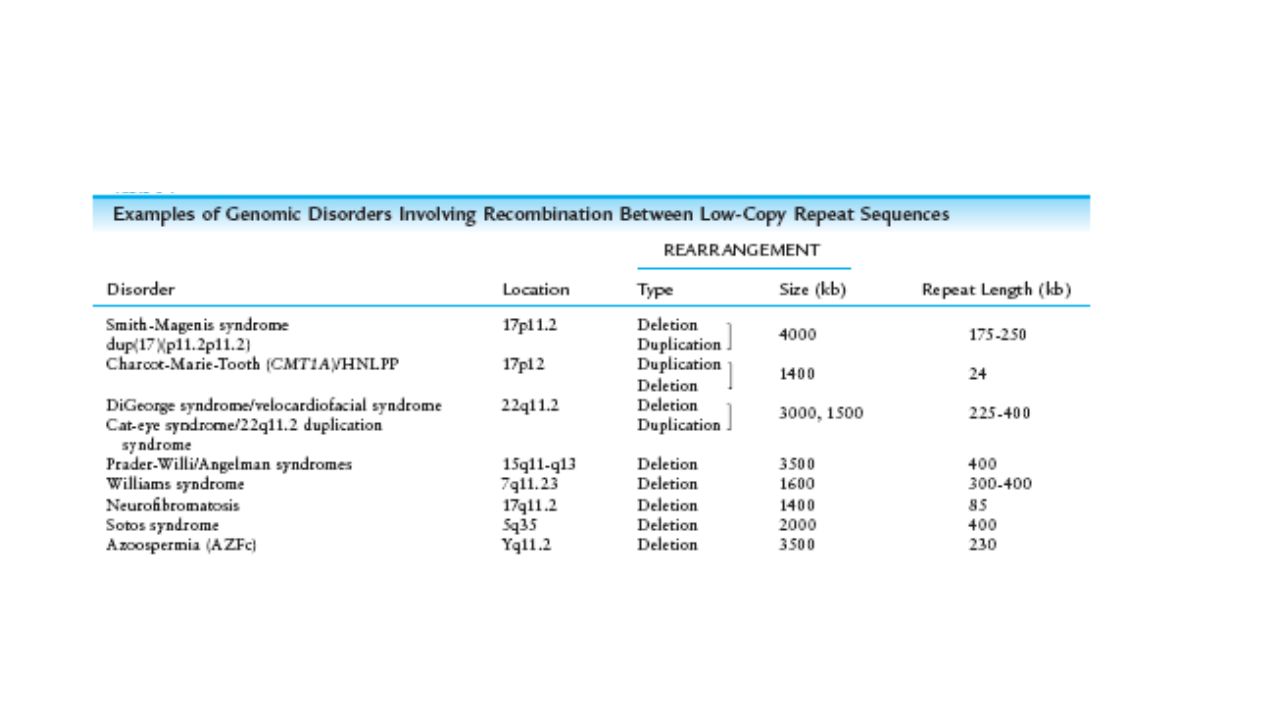
Mikrodelesyon Sendromları
Prader Willi Sendromu Angelman Sendromu Williams Sendromu 22q11 delesyon sendromları (VCFS,Di-George sendromu) Smith Magenis Sendromu Miller-Dieker Sendromu
 Smith Magenis Sendromu. Miller-Dieker Sendromu.")
11
22q11 delesyon sendromları
Mikrodelesyon sendromlarından Velokardiofasiyal sendromu,Di George sendromu ve Conotrunkal yüz anomalisi farklı fenotipik bulgulara sahip aynı genetik defekte sahip klinik durumu gösterir. Mikrodelesyonun boyutları ve gen içeriği fenotipin şiddetini belirler. 22q11 delesyon sendromları tanısı FISH yöntemi kullanılarak konur.

13
Cri Du Cat Sendromu Cri du cat sendromunda ağlarken kedi sesi çıkarılması tanı koydurucudur. 5p del. Sendromu olarak bilinen hipertelorism, retrognatia ve tipik ağlama sesi ile karakterizedir. Kromozom 5p nin delesyonu FISH ile gösterilmesi ile kesin teşhis konur.

14
Nadir Hastalıklar-Genetik-I
Avrupa’da 1/2000 görülen hastalıklar nadir kabul edilir. Bilinen binlerce nadir hastalık vardır. Akdeniz bölgesinde görülen bir talasemi burda yaygın iken Karadeniz’de nadir görülür. Nadir hastalıkların büyük çoğunluğu genetik nedenlerle oluşur. Yarısına yakını erişkin yaş grubunda olur. Nadir hastalıklar ciddi, kronik ve ilerleyicidir. Nadir hastalık alanında tıbbi ve bilimsel bilgi açığı vardır. Çoğu nadir hastalığı tam iyileştiren bir tedavi olmamasına karşın uygun tıbbi girişim ve bakım yaşam kalitesini arttırır.

15
Nadir Hastalıklar-Genetik-II
Bu hastalıkların teşhis ve tedavisinde gün geçtikçe yenilikler son hızla devam etmektedir. Günümüzde nadir hastalıkların tanısı moleküler genetik testlerle konulabilmektedir. Nadir hastalıklara ait arşivlerin oluşturulması ve doğal öyküleri hakkında bilgi edinilebilmiştir. Birçok Avrupa ülkesinde nadir hastalıklara ait ulusal politalarla sunulan bakış açıları sayesinde yeni umutlar doğmaktadır. Nadir veya ‘’orphan’’ hastalıklar envanteri ve 6000’den fazla hastalık hakkında bilgi ve aynı zamanda Avrupa’da uzmanlaşmış hizmetler rehberi sunmaktadır.

16
Nadir Hastalıkların Tedavisi
Orphan drug veya Yetim ilaç diye bilinen çok nadir hastalıklar için üretilen küçük Pazar payına sahip ve ilaç endüstrisinin üretmekte gönülsüz olduğu bir gruptur. AB tarafından düzenlenen yetim ilaç teşvik tedbirleri tanımı, Nadir hastalıklar uzman komitesi da yayımlanmıştır.Her ay güncellenen yetim ilaçlar listesi güncellenmektedir. Nadir hastalıklar cenneti olan ülkemizde maalesef henüz bu konuda yeterli girişim ve düzenleme bulunmamaktadır. Orphanet websitesinden yetim ilaç statüsündeki ürün adı, madde adı,sponsor adı ve ülke adına ulaşılabilmektedir.

17
Orphanet Orphanet Avrupa komisyonu tarafından 6 ayrı dilde hizmet veren,herkesin kullanımına açık internet portalıdır. 2007 yılında kurulan orphanet-Türkiye, nadir hastalıklar ve yetim ilaçlar konusunda genel bilinci arttırmak ve Ulusal veri tabanlarının tanıtıp kullanılmasını sağlamayı hedefler. Orphanet,nadir hastalık alanında tanı ve tedavi olanaklarının iyileştirilmesine,bilimsel ve klinik araştırmaların artırılmasına, yaşam kalitesinin yükseltilmesine, kaynakların daha verimli kullanılmasını amaçlar. Nadir hastalıklarla ilgilenen uzman doktorlar, tanı ve tedavi üzerine uzmanlaşan klinikler, laboratuvar ve hastaneler, yetim ilaçlar ile uğraşan ilaç firmaları, hasta organizasyonları ve araştırma grublarına ait Uluslararası Veri tabanına kayıt oluşturur.


>")
>")

















