Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Nefroloji Bilim Dalı Olgu Sunumu 29 Nisan 2015 Çarşamba Dr. Didem Durak

2
8 yaş 9 aylık kız hasta

3
Hasta 28 Aralık tarihinde ateş yüksekliği şikayeti ile dış merkeze başvurmuş. Ateş dışında şikayet yok.

4
Yapılan tetkiklerinde T İ T ‘de Dansite:1012 Eser lökosit (mik. 38) +3 kan (mik.278) +3 protein Wbc:19,100 u/l Neu:16,300 u/l Hgb:13,4 g/dl Plt:269.000 u/l Üre:9,4 mg/dl Cre:0,75 mg/dl AST:20 IU/l ALT:11 U/l CRP:5,8 mg/dl Albumin:3,2 g/dl TG:69 HDL:38 Tot.kolestrol:191
 +3 kan (mik.278) +3 protein Wbc:19,100 u/l Neu:16,300 u/l Hgb:13,4 g/dl Plt: u/l Üre:9,4 mg/dl Cre:0,75 mg/dl AST:20 IU/l ALT:11 U/l CRP:5,8 mg/dl Albumin:3,2 g/dl TG:69 HDL:38 Tot.kolestrol:191.")
5
Hasta pyelonefrit tanısı ile yatırılmış. Seftriakson tedavisi başlanmış. Üriner USG yapılmış. Her iki böbrekte grade 1 eko artışı. Her iki böbrek boyut, parenkim kalınlı ğ ı normal. Mesane konturları düzenli. Alınan idrar kültüründe üreme olmamış. (antibiyotik sonrası?)
.")
6
28.12.1429.12.1402.01.1505.01.15 Dansite 1012 +3 protein +2 protein +3 kan +1 kanKan negatif Eser lökosit+1 lökositLökosit neg.

7
Hasta proteinürisinin devam etmesi üzerine yönlendirildi.

8
Fizik Muayane Tartı:25,4 kg (25-50p) Boy:127 cm ( 5-25p) TA:115/59 mmhg (95th 125/76 mmhg) Genel durumu iyi Cilt turgor tonus do ğ al, ödem yok. Orofarinks tonsiller do ğ al. Solunum sesleri do ğ al,bilateral eşit, ral yok. Sternum üzerinde operasyon skarı S1 + S2 + üfürüm yok. Batın rahat HSM yok asit yok.

9
Özgeçmiş Miadında NSVY do ğ um 1 yaşında VSD opere

10
Soygeçmiş Anne 38 yaş sa ğ sa ğ lıklı Baba 42 yaş sa ğ sa ğ lıklı, DM Akrabalık yok. 1.çocuk 21 yaş erkek sa ğ sa ğ lıklı 2. çocuk 19 yaş erkek sa ğ sa ğ lıklı 3. çocuk hastamız Anne tarasında Alport sendromu

11
Yapılan tetkiklerinde 06.01.15 T İ T ‘de Dansite: +1 kan (mik.278) +1 protein Pro/cre:3 ASO:187 C3:138 (N) C4:30,8 (N) Üre:mg/dl Cre:0,68 mg/dl AST:20 IU/l ALT:11 U/l CRP:5,8 mg/dl Albumin:3,2 g/dl TG:233 HDL:18 Tot.kolestrol:180 LDL:115
 +1 protein Pro/cre:3 ASO:187 C3:138 (N) C4:30,8 (N) Üre:mg/dl Cre:0,68 mg/dl AST:20 IU/l ALT:11 U/l CRP:5,8 mg/dl Albumin:3,2 g/dl TG:233 HDL:18 Tot.kolestrol:180 LDL:115")
12
07.01.15 pro/cre:2,2

13
13.01.15 T İ T: 24 saat idrar protein:77mg/m2/st İ drar sedimenti: 2-3 eritrosit 27.01.15 24 saat idrar protein:80mg/m2/st İ drar sedimenti temiz ASO, C3, C4 normal TA 111/78 mmhg

14
03.03.15 24 saat idrar protein:112 mg/m2/st İ drar sedimenti temiz Göz muayenesi ve işitme testi normal. TA 100/63 mm hg

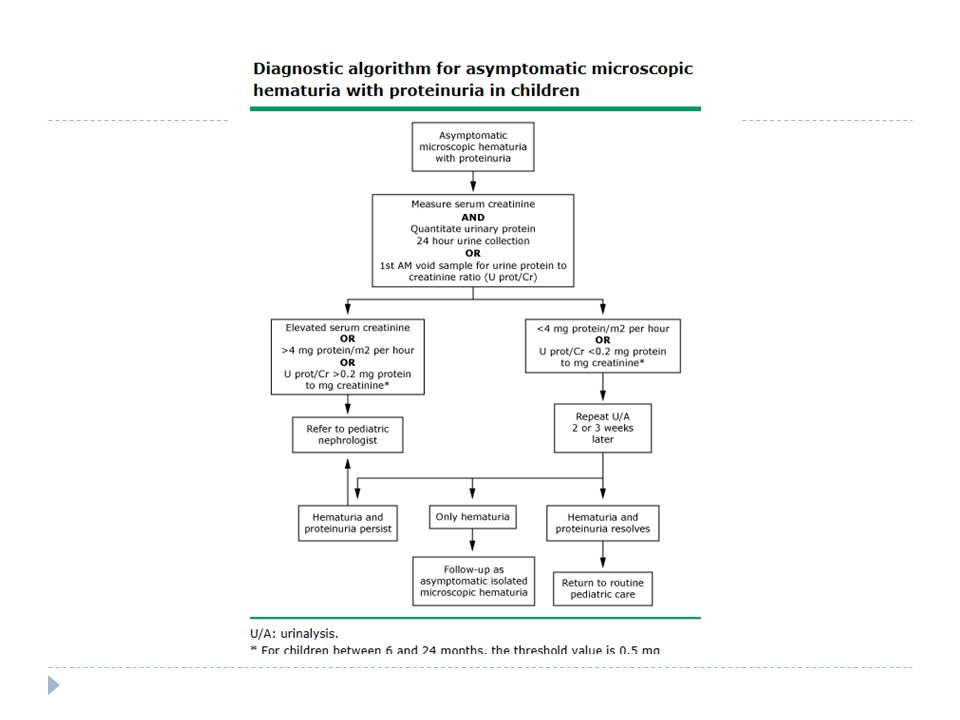
16
Ön tanılar?

17
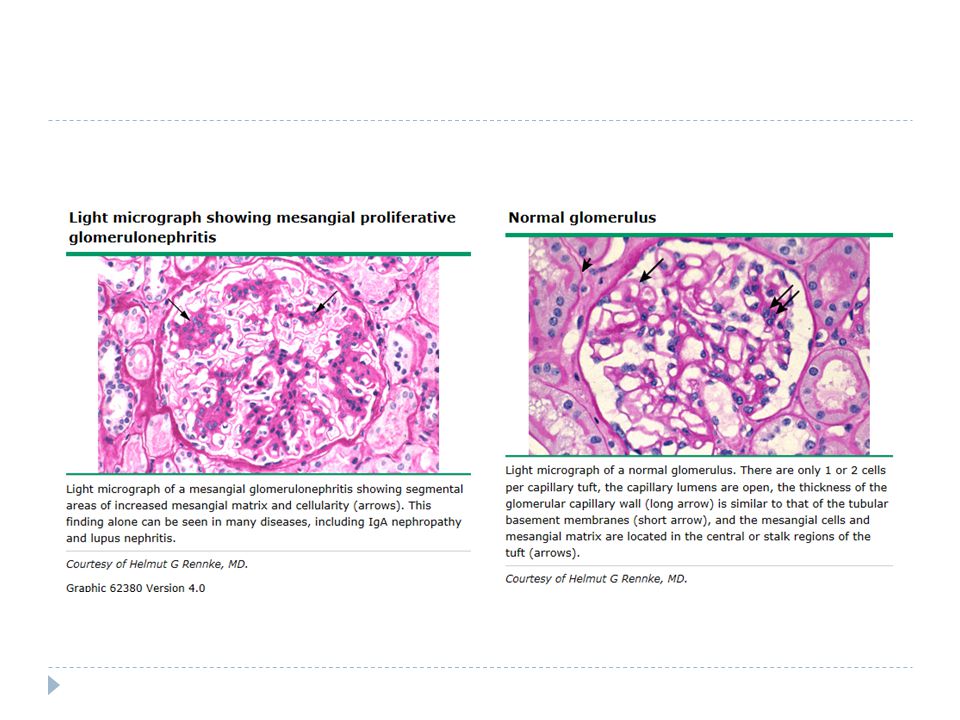
25.03.15 Böbrek Biopsisi

19
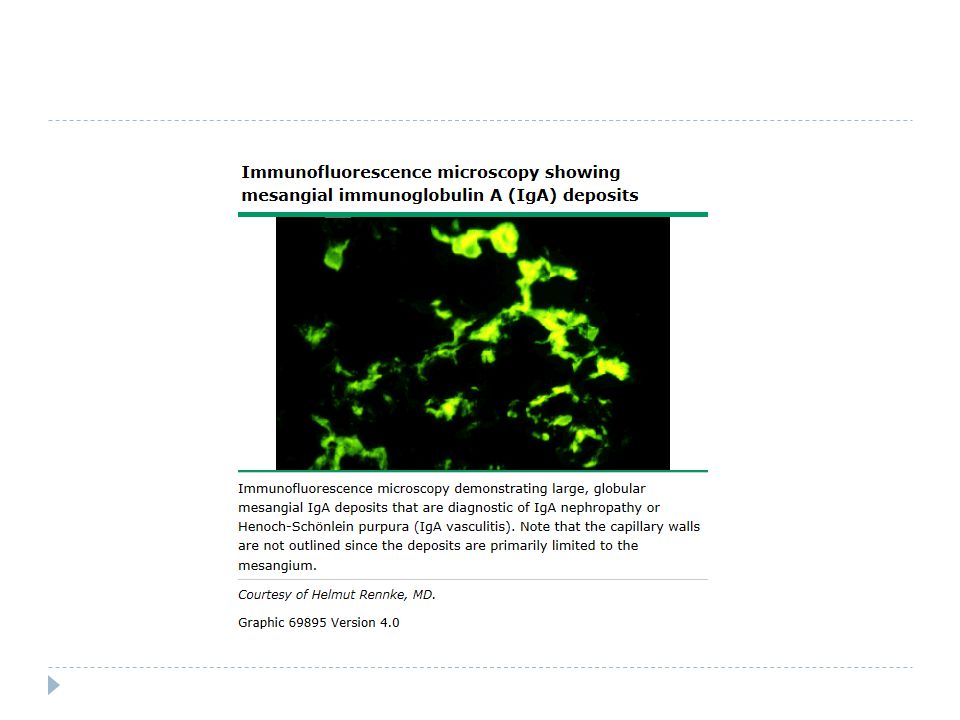
Ig A nefropatisi çocukluk ça ğ ında en sık nefropati

20
Klinik Viral ÜSYE veya bakteriyel tonsillit sonrası tekrarlayan makroskopik hematüri Rutin kontrolde mikroskopik hematüri ve nefrotik düzeyde olmayan proteinüri Nefrotik sendrom / hızlı gidişli GN

21
Patoloji Tanı patoloji ile konur.

22
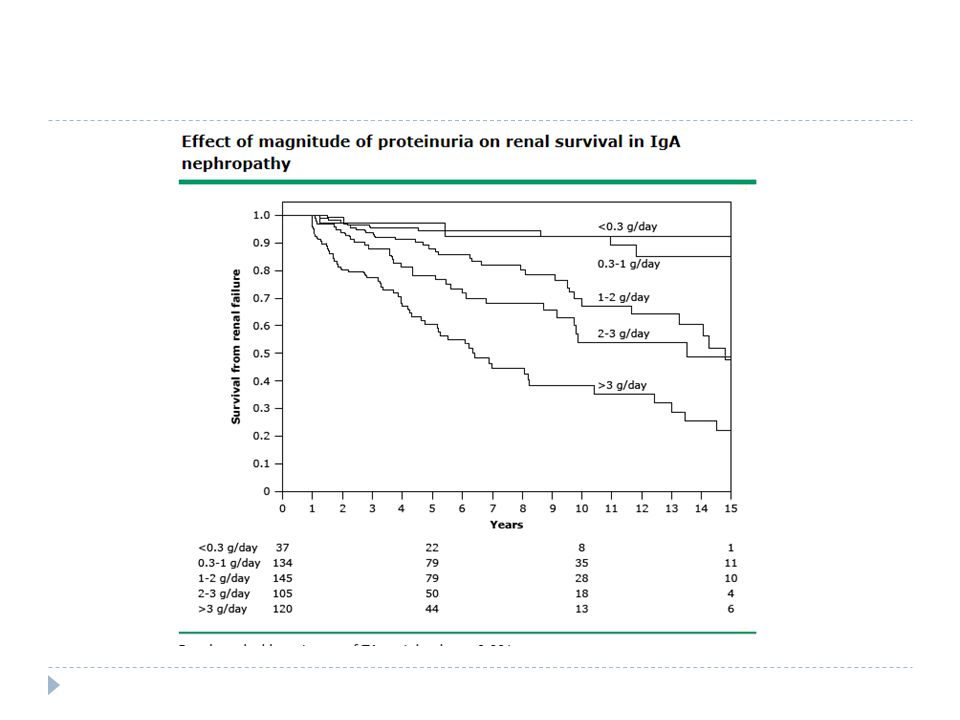
Prognoz Proteinüri eşlik etmedi ğ i veya çok az oldu ğ u hastalar düşük riskli Serum kreatitin yüksekli ğ i Hipertansiyon Proteinüri varsa progresiv hastalık riski fazla

23
14.04.15 kontrol Şikayet yok FM ödem yok, ek özellik yok TA 112/91 mm hg T İ T +3 protein +1 kan İ drar sedimenti 1-2 eritrosit Delix ve balık ya ğ ı başlandı. Tuz kısıtlama

25
Patients with IgA nephropathy who have little or no proteinuria (less than 500 to 1000 mg/day) usually have a low risk of progression. However, progressive proteinuria and renal insufficiency develop in a substantial proportion of patients over the long term [7-12]. Among patients who develop overt proteinuria and/or an elevated serum creatinine concentration, progression to end-stage renal disease is approximately 15 to 25 percent at 10 years and 20 to 30 percent at 20 years [3,4,11-14].7-123,4,11-14

26
CLINICAL FEATURES — Patients with IgA nephropathy typically present in one of three ways; the relative frequency depends in part upon screening practices (which will lead to increased discovery of asymptomatic cases) and the particular population being evaluated [4,5]:4,5 ● Approximately 40 to 50 percent present with one or recurrent episodes of visible hematuria, usually following an upper respiratory infection. This has sometimes been called "synpharyngitic hematuria." These episodes can be provoked by bacterial tonsillitis, or by other viral upper respiratory infections; they may occur in individuals who have already undergone tonsillectomy. It is presumed, although not proven, that the first episode represents the onset of the disease. Patients may complain of flank pain during acute episodes, which usually reflects stretching of the renal capsules. Low-grade fever may also be present. These features can mimic urinary tract infection or urolithiasis. Most patients have only a few episodes of visible hematuria and episodes usually recur for a few years at most. A discussion of clinical clues that may distinguish IgA nephropathy from poststreptococcal glomerulonephritis can be found separately. (See "Differential diagnosis and evaluation of glomerular disease", section on 'Hematuria following upper respiratory infection'.)"Differential diagnosis and evaluation of glomerular disease", section on 'Hematuria following upper respiratory infection' ● Another 30 to 40 percent have microscopic hematuria and usually mild proteinuria, and are incidentally detected on a routine examination [21,22]. In these patients, the disease is of uncertain duration. Gross hematuria will eventually occur in 20 to 25 percent of these patients.21,22 ● Less than 10 percent present with either nephrotic syndrome or acute rapidly progressive glomerulonephritis picture characterized by edema, hypertension, and renal insufficiency as well as hematuria. Rarely, IgA nephropathy may present with malignant hypertension. It is usually presumed that patients have longstanding disease which was not detected earlier because the patient did not have visible hematuria or undergo routine urinalysis.
![ CLINICAL FEATURES — Patients with IgA nephropathy typically present in one of three ways; the relative frequency depends in part upon screening practices (which will lead to increased discovery of asymptomatic cases) and the particular population being evaluated [4,5]:4,5 ● Approximately 40 to 50 percent present with one or recurrent episodes of visible hematuria, usually following an upper respiratory infection.](http://images.slideplayer.biz.tr/16/5251747/slides/slide_26.jpg "This has sometimes been called synpharyngitic hematuria. These episodes can be provoked by bacterial tonsillitis, or by other viral upper respiratory infections; they may occur in individuals who have already undergone tonsillectomy. It is presumed, although not proven, that the first episode represents the onset of the disease. Patients may complain of flank pain during acute episodes, which usually reflects stretching of the renal capsules. Low-grade fever may also be present. These features can mimic urinary tract infection or urolithiasis. Most patients have only a few episodes of visible hematuria and episodes usually recur for a few years at most. A discussion of clinical clues that may distinguish IgA nephropathy from poststreptococcal glomerulonephritis can be found separately. (See Differential diagnosis and evaluation of glomerular disease , section on Hematuria following upper respiratory infection .) Differential diagnosis and evaluation of glomerular disease , section on Hematuria following upper respiratory infection ● Another 30 to 40 percent have microscopic hematuria and usually mild proteinuria, and are incidentally detected on a routine examination [21,22]. In these patients, the disease is of uncertain duration. Gross hematuria will eventually occur in 20 to 25 percent of these patients.21,22 ● Less than 10 percent present with either nephrotic syndrome or acute rapidly progressive glomerulonephritis picture characterized by edema, hypertension, and renal insufficiency as well as hematuria. Rarely, IgA nephropathy may present with malignant hypertension. It is usually presumed that patients have longstanding disease which was not detected earlier because the patient did not have visible hematuria or undergo routine urinalysis..")
33
ETIOLOGY — The etiology of IgA nephropathy is unknown in the great majority of cases. The possibility that infections contribute to the underlying pathogenesis of this disorder has been explored, as have genetic associations. It has also been suggested that IgA nephropathy results from hypersensitivity to food antigens, in view of its association with celiac disease. There is, however, no evidence for widespread hypersensitivity to food antigens in IgA nephropathy [85].85 In our opinion, IgA nephropathy is an autoimmune disease resulting from dysregulation of mucosal-type IgA immune responses. The autoantigens are a specific set of IgA1 O-glycoforms displaying poor O-linked galactosylation of the IgA1 hinge region. These O-glycoforms result in the generation of hinge glycan-specific IgA and IgG autoantibodies in susceptible individuals. As a result, any mucosal infection or food antigen may drive the production and release of pathogenic IgA into the circulation where it has the propensity to deposit within the mesangium and trigger glomerular injury. Why patients with IgA nephropathy are predisposed to high levels of these IgA1 O-glycoform autoantigens remains unknown.

Benzer bir sunumlar