Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Nöroloji Bilim Dalı Olgu Sunumu 22 Aralık 2015 Salı Yandal Ar. Gör. Uzm. Dr. Ayfer Sakarya Güneş

2
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Çocuk Nöroloji B.D. Olgu Sunumu 22.12.2015 Dr. Ayfer Sakarya Güneş

3
4,5 yaşında kız hasta Şikayeti : Konuşma geriliği, dengesizlik, ayakta dik duramama

4
12 aylık yürümeye başlayan hastanın parmak ucu yürümesi ve dengesizliği varmış. Zeynep Kamil EAH’de yapılan tetkiklerinde B12 vitamin eksikliği saptanmış. Özel eğitim ve fizik tedavi desteği alıyor. Hikaye

5
Prenatal: Annenin 3. gebeliği. Gebeliği boyunca düzenli doktor kontrolü ve ultrason kontrolü yok.Gebelik sırasında sigara,alkol,madde kullanımı,kanama,akıntı,radyasyon maruziyeti,idrar yolu enfeksiyonu, döküntülü ya da ateşli hastalık geçirme öyküsü yok. Natal: Hastanede, miadında, sezaryenle, 3250 gr olarak doğmuş. Postnatal: Doğar doğmaz ağlamış.Küvez bakımı almamış.İkter, siyanoz öyküsü yok. Beslenme: İlk 6 ay yalnızca anne sütü almış. Daha sonra ek gıdalara geçilmiş.Toplam 1 yıl anne sütü almış. Büyüme- Gelişme: Yaşıtlarıyla uyumlu seyretmiş. 12 aylıkken desteksiz yürümüş, ancak parmak ucu basıyor ve dengesini sağlayamıyormuş. Aşılar: Sağlık ocağı aşıları tam Geçirdiği hastalıklar: MTHFR C677T heterozigot mutasyonu olması nedeni ile 1 yıldır folbiol 1x1/4 tb kullanıyor. (Göztepe EAH Metabolizma başlamış) Allerji: Özellik yok Özgeçmiş
 Allerji: Özellik yok Özgeçmiş.")
6
Anne: 35 yaşında,İO mezunu, evhanımı, sağ- sağlıklı Baba: 35 yaşında, ortaokul mezunu, sağ-ülseratif kolit Anne ve baba arasında akrabalık var. Anne ile babanın dedeleri kardeş. 1. çocuk: Erkek, 7 yaşında, sağ-sağlıklı 2. çocuk: Hastamız 3.çocuk:Erkek, 2 yaşında, sağ-sağlıklı Ailede sürekli hastalık: Yok Soygeçmiş

7
Ateş: 36.8°C Nabız: 98/dak Tansiyon: 116/75 mm Hg Solunum: 24/dak Boy: cm (p) Kilo: kg (p) Fizik Muayene
 Kilo: kg (p) Fizik Muayene")
8
Genel durum: iyi Cilt: Turgor-tonus doğal. Baş-boyun: Saç ve saçlı deri doğal. Kafa yapısı simetrik. Boyunda kitle ve LAP yok. Gözler: Işık refleksi bilateral mevcut. Pupiller izokorik. Konjonktivalar ve skleralar doğal. Göz kürelerinin her yöne hareketi doğal.Heliotrop raş yok. Kulak-Burun-Boğaz: Bilateral kulak zarları doğal. Burun tıkanıklığı,akıntısı yok. Kardiyovasküler: S1-S2 doğal. Üfürüm yok.S3 yok. AFN her iki alt ekstremitede alınıyor. Kalp tepe atımı 5. interkostal aralıkta. Solunum sistemi: Her iki hemitoraks solunuma eşit katılıyor. Ral, ronküs, ekspiryum uzunluğu yok. Fizik Muayene

9
Gastrointestinal sistem: Batın rahat. Hassasiyet yok.Defans,rebound yok. Hepatomegali, splenomegali yok. Barsak sesleri doğal. Genitoüriner sistem: Haricen erkek, anomali yok. Nöromusküler sistem: Bilinç açık, koopere –oryante. Kranial sinir muayenesi doğal. Patolojik refleks yok. Alt ve üst ekstremite DTR’ler alınamadı. Parmak ucu ve geniş tabanlı yürümesi var. Ekstremiteler: Kas kitlesi doğal, deformite yok. Başını dik tutabiliyor, desteksiz oturabiliyor, göz takibi var, seslenince ismine bakıyor. Fizik Muayene

10
Laboratuvar

11
Ön Tanı?

12
Serebellum ve Beyin Sapı Herediter Dejeneratif Hastalıkları Serebellar lezyon veya fonksiyon bozukluğunda; kas tonusu regülasyonu, motor kontrol ve hareketlerin koordinasyonu ile ilgili sorunlar ortaya çıkar. Gelişimsel anomali, dejeneratif hastalık, vasküler olaylar ve travma gibi nedenlere bağlı serebellum yapısında veya ileti sistemindeki bozukluklar, karakteristik motor belirti ve bulguların ortaya çıkmasıyla sonuçlanır.

13
Serebellum ve Beyin Sapı Herediter Dejeneratif Hastalıkları Serebellum ve/veya beyin sapının etkilendiği herediter dejeneratif hastalıklar genellikle yavaş fakat ilerleyici bir klinik tabloya neden olurlar. Herediter ataksilerin klinik olarak sınıflandırılmasında birtakım güçlükler bulunmaktadır. Genetik tanı ile ilgili teknolojiler gelişmeden önce, bu hastalıklar kalıtım tipi, klinik özellikleri ve patolojik bulgulara dayanarak sınıflandırılmıştır.

14
Ancak, bu yaklaşımların yetersiz olduğu ve genellikle tanı ile ilgili netlikten çok daha fazla kafa karışıklığına yol açtığı kanıtlanmıştır. Son 20 yılda genetik ve moleküler biyoloji tekniklerinin yaygın kullanımı ile kalıtsal ataksilerin patolojik mekanizmaları ve olası tedavi yöntemleri hakkında fikir sağlayacak, hassas bir sınıflandırma şeması yapılmıştır.

15
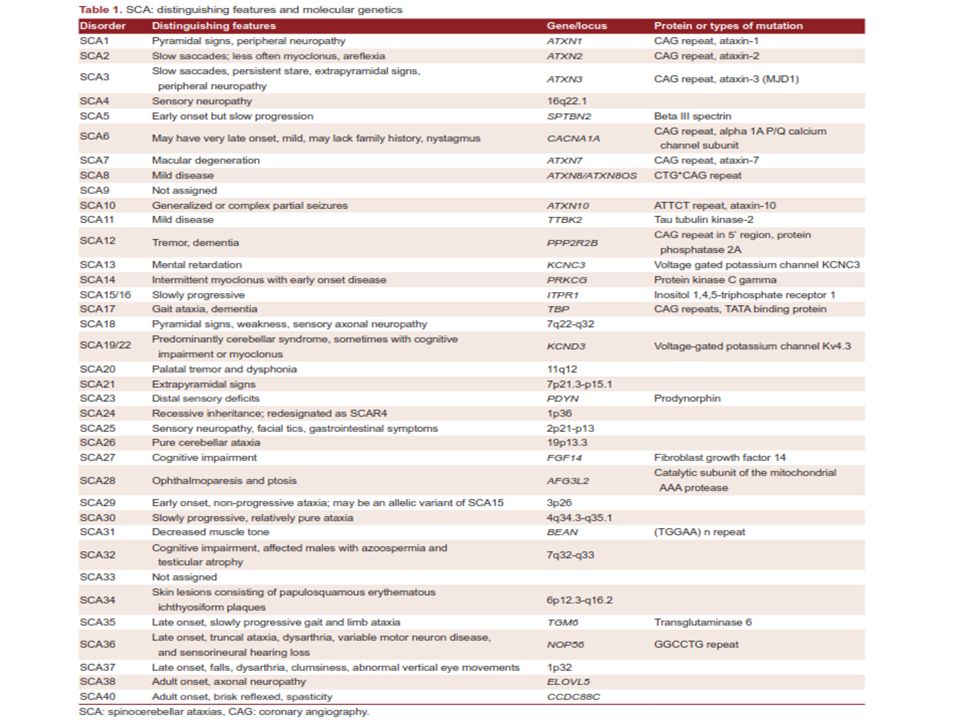
Herediter ataksiler kalıtım tipi ve tanımlanan genetik defekte göre; -otozomal dominant -otozomal resesif -X’e bağlı geçiş -maternal kalıtım(Mitokondriyel) olarak dört alt sınıfa ayrılmıştır.
 olarak dört alt sınıfa ayrılmıştır.")
19
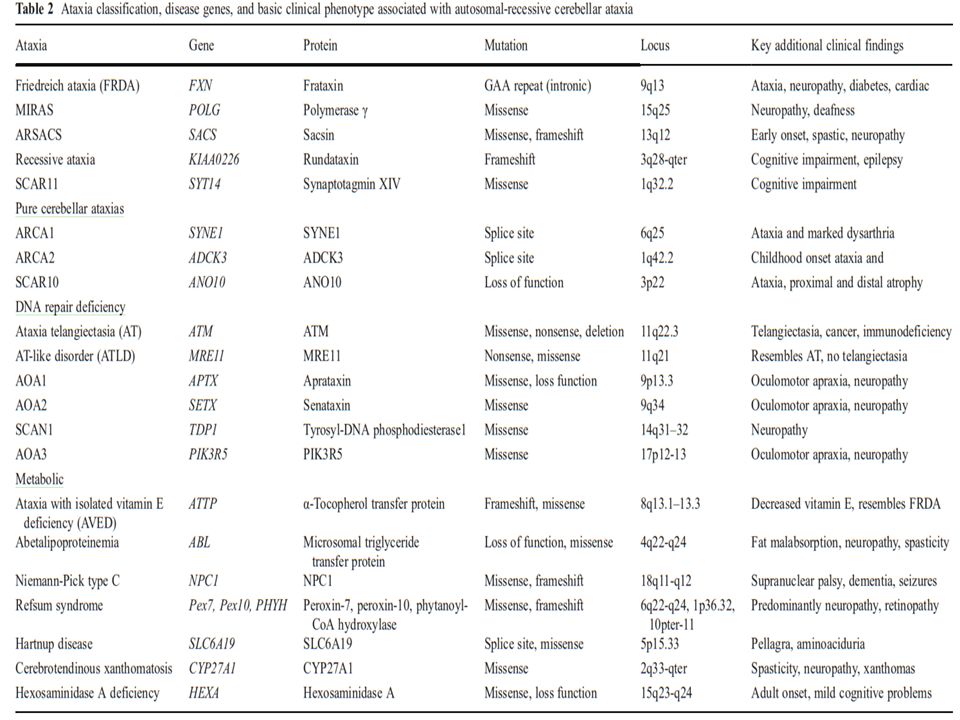
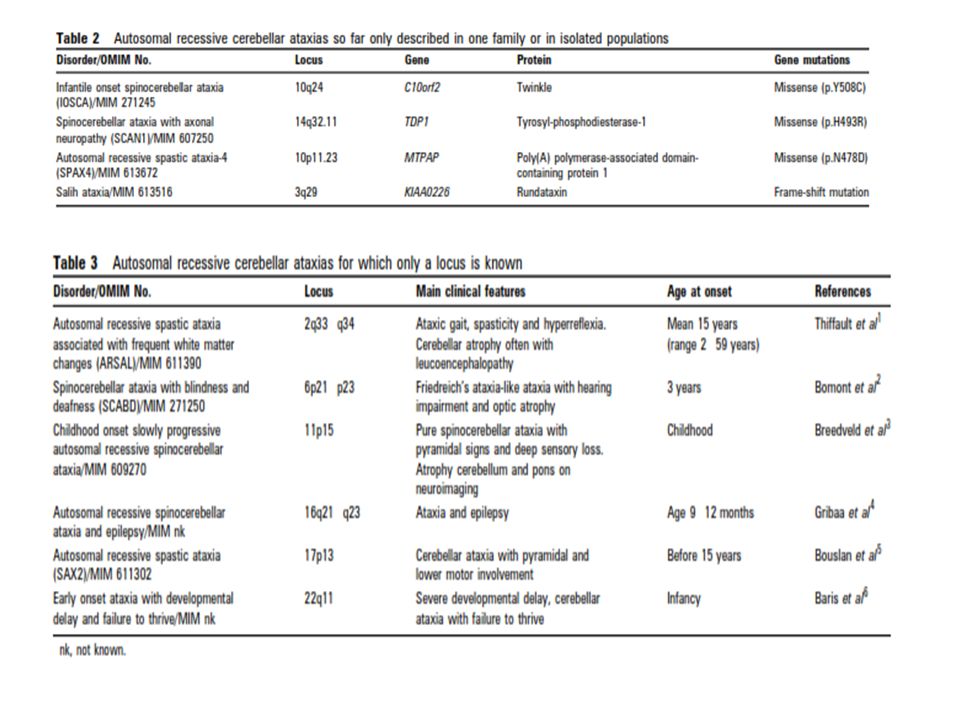
Otozomal Resesif Serebellar Ataksiler (ARCAs) Herediter ataksilerin klinik ve genetik olarak oldukça heterojen özellikler taşıması, bu hastalıkların teşhis ve tanısal yaklaşımını oldukça zorlaştırmaktadır. OR serebellar ataksilerde progresif serebellar ataksi halen en belirgin ve değişmez özelliktir. Sıklıkla, diğer nörolojik ve nörolojik olmayan semptomlar da eşlik etmektedir. Genellikle infant veya erken çocukluk döneminde başlama eğilimleri vardır. Sıklık, yaklaşık olarak 2,2-7/100000 (toplumlara göre değişmektedir)
.")
20
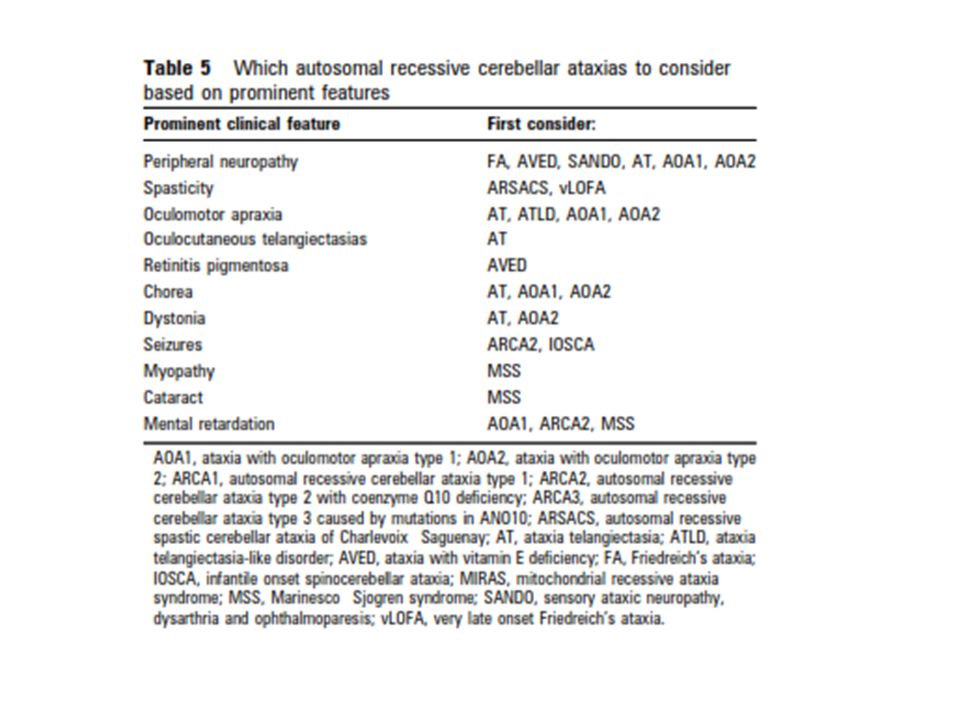
Ataksi-Okulomotor Apraksi Tip 1 (AOA 1) İlk kez 1971 yılında Japonya’da bildirilmiştir. Japonya’da en sık görülen OR Serebellar Ataksi tipidir. Avrupa ve Kuzey Afrika’da da sık görülmektedir. Klinik bulgular genellikle 7 yaşından sonra ortaya çıkar. Ancak daha küçük yaşlarda da (2- 5 yaş) başlayabilir.
 başlayabilir..")
21
İlerleyici serebellar ataksi ve disartri tipik başlangıç bulgularıdır. Ancak bazen kore başlangıç bulgusu olabilir. Okuler apraksi, tipik olarak semptomların başlangıcından birkaç yıl sonra ortaya çıkar. Zaman içinde progresif oftalmopleji tablosu oturur. (Ayırıcı özelliktir, %80 hastada görülür) Duyusal ve motor nöropati OMA ve nöropati başlangıçta genellikle yoktur, ancak zaman içinde şiddetli nöropati fenotipteki baskın özellik haline gelmektedir.
 Duyusal ve motor nöropati OMA ve nöropati başlangıçta genellikle yoktur, ancak zaman içinde şiddetli nöropati fenotipteki baskın özellik haline gelmektedir..")
22
Eşlik edebilen diğer klinik bulgular; -arefleksi (alt ekstremitede) -distoni -koreatetoz -retinal/maküler lezyonlar -bilişsel gerilik( şiddetli zeka geriliği/normal aralığında olabillir) Hastalar tanıdan itibaren, ortalama 10 yıl içerisinde yürüme yetilerini kaybederler. (5-20 yıl) Hastalık başlangıcından yaklaşık 10-15 yıl sonra %80 hastada hipoalbuminemi ve hiperkolesterolemi görülmektedir. Yaşam süresi etkilenmemiştir.
 Hastalık başlangıcından yaklaşık yıl sonra %80 hastada hipoalbuminemi ve hiperkolesterolemi görülmektedir. Yaşam süresi etkilenmemiştir..")
23
AOA 1, 9p13.3 kromosomunda bulnan APTX gen mutasyonuna bağlı ortaya çıkar. APTX geni, uzun ve kısa olmak üzere, iki izoformu bulunan aprataksin proteini kodlar. Uzun izoform(356 aa) 3 fonksiyonel bölüm içermektedir; *N-terminal (forkhead associated=FHA) *Santral ( Histidin triad=HIT) *C-terminal (Zing Finger=ZNF) Mutasyonların çoğunluğu HIT bölümündedir.
 3 fonksiyonel bölüm içermektedir; *N-terminal (forkhead associated=FHA) *Santral ( Histidin triad=HIT) *C-terminal (Zing Finger=ZNF) Mutasyonların çoğunluğu HIT bölümündedir..")
24
FHA; -X-ray repair cross-complemanting protein 1 (XRCC1) ile DNA ligaz IIIα kompleksinin oluşmasına - X-ray repair cross-complemanting protein 4 (XRCC4) ile DNA ligaz 4 kompleksinin oluşmasına aracılık eder. Bu etkileşimler APTX geninin DNA’daki tek zincir ve çift zincir kırık onarımında rol oynadığını desteklemektedir.

25
FHA: forkhead associated; NLS: nuclear localization signal; HIT: Histidin triad; ZNF: Zinc finger

26
AOA 1 hastalarındaki etkilenmiş hücreler, zincir kırıklarını tetikleyen ajanlara karşı oldukça hassas hale gelirler. Oksidatif stres varlığında özellikle nöronal hücre ölümüne neden olan zincir kırık kümeleri oluşmaktadır. Son çalışmalarda aprataksinin mitokondrilerde lokalize olduğu ve mitokondriyel fonksiyonun korunmasında da rol oynadığı ileri sürülmektedir. Patolojik bulgular, serebellar purkinje hücrelerinde kayıp; arka kolon, spinosrebellar yol ve ön boynuz hücrelerinde dejenerasyondur.

27
Nörogörüntüleme; serebellar, özellikle vermiste atrofi görülür. Bazı hastalarda beyin sapı atrofisi de bildirilmiştir. Laboratuar; hastalığın geç dönemlerinde hipoalbuminemi ve hiperkolesterolemi görülebilir. AFP düzeyi normaldir.

28
Tanı; klinik şüphe ve mutasyon analizi Tedavi; Koenzim Q10

29
Ataksi Okulomotor Apraksi Tip 2 (AOA 2) İlk kez 1988 yılında 4 Japon hastada tanımlanmış. 3-30 yaş aralığında(sıklıkla13-19 yaş) başlayan, aksonal duyusal-motor nöropatinin eşlik ettiği serebellar ataksi ile karakterizedir. Okulomotor apraksi hastaların yaklaşık yarısında görülür. (Bir çalışmada içe şaşılık oranı OMA’den daha yüksek bulunmuş) Ellerde distonik postür, koreiform hareketler, baş veya postural el tremoru görülebilir. Hareket bozuklukları, AOA 1 aksine, genellikle persiste eder, zaman içinde kaybolmaz.
 başlayan, aksonal duyusal-motor nöropatinin eşlik ettiği serebellar ataksi ile karakterizedir. Okulomotor apraksi hastaların yaklaşık yarısında görülür. (Bir çalışmada içe şaşılık oranı OMA’den daha yüksek bulunmuş) Ellerde distonik postür, koreiform hareketler, baş veya postural el tremoru görülebilir. Hareket bozuklukları, AOA 1 aksine, genellikle persiste eder, zaman içinde kaybolmaz..")
30
Ataksi Okulomotor Apraksi Tip 2 (AOA 2) Bilişsel fonksiyonlar genellikle normaldir. Albumin değeri normal, AFP değeri yüksektir. Kranyal MR’da serebellar atrofi vardır. AOA 1’den ayırt ettirici özellikler; *Daha geç başlangıç yaşı *Normal albumin değeri *Artmış AFP değeri AOA 2, kromozom 9q34’de gösterilen, sintaksin proteinini kodlayan SETX gen mutasyonuna bağlı gelişir.

31
Freidreich Ataksi (FRDA) FRDA, en sık görülen herediter ataksidir. Başlangıç yaşı genellikle puberte dönemi olup, ilk bulgu ilerleyici ataksidir (ekstremite ve yürüyüş). Diğer klinik bulgular; * Alt ekstremitede DTR alınamaz. (zamanla üst ekst. de alınamaz) *Aksonal duyu nöropati (ilk 5 yıl içinde), distallerde pozsiyon ve vibrasyon duyu kaybı *Disartri, disfaji *Ekstensor plantar yanıt, * Hastalığın geç döneminde alt ekstremitede pramidal güç kaybı ve spastik, hiperrefleksi gelişebilir
. Diğer klinik bulgular; * Alt ekstremitede DTR alınamaz. (zamanla üst ekst. de alınamaz) *Aksonal duyu nöropati (ilk 5 yıl içinde), distallerde pozsiyon ve vibrasyon duyu kaybı *Disartri, disfaji *Ekstensor plantar yanıt, * Hastalığın geç döneminde alt ekstremitede pramidal güç kaybı ve spastik, hiperrefleksi gelişebilir.")
32
Freidreich Ataksi (FRDA) Nörolojik sistem dışında görülebilen diğer bulgular; *katart, nistagmus, retinitis pigmentosa, optik atrofi(%25) *Sensörinöral işitme kaybı *kardiyomiyopati (ilerleyicidir ve kalp yetmezliği nedeni ile en önemli mortalite nedenidir) *pes kavus, kifoskolyoz *Diyabetes mellitus (insülin direci??)
 Nörolojik sistem dışında görülebilen diğer bulgular; *katart, nistagmus, retinitis pigmentosa, optik atrofi(%25) *Sensörinöral işitme kaybı *kardiyomiyopati (ilerleyicidir ve kalp yetmezliği nedeni ile en önemli mortalite nedenidir) *pes kavus, kifoskolyoz *Diyabetes mellitus (insülin direci )")
33
Freidreich Ataksi (FRDA) Kromozom 9q13’de yer alan X25 geni, frataksin adlı bir mitokondriyal proteini kodlar. Frataksin iç mitokondriyal membranda bulunan bir proteindir. Mitokondriyal solunum ve demir hemostazında rol oynar. Fratansin eksikliği mitokondriyal demir birikimi, fonksiyonlarında bozulma, oksidadif strese karşı duyarlılık oluşur. Serbest radikal birikimi ve hücre ölümü ile sonuçlanır. Tedavide antioksidanlar kullanılmaktadır. Koenzim Q10 ve E vit. takviyesi ile nörolojik ve kardiyak bulgularda iyileşme olduğu bildirilmiş. Koenzim Q10 analoğu olan idebenone kullanımı ile kardiyak bulguların iyileştiği ancak nörolojik bulgularda düzelme olmadığı gösterilmiş.

34
Ataksi Telenjektazi (AT) OR geçişli herediter ataksiler arasında, FRDA’ den sonra ikinci sıklıkta görülmektedir. Serebellar ataksi, okulokutanöz telenjektazi, bağışıklık sistemi bozukluğu ve melignensi eğilimi en belirgin özellikleridir. 2-3 yaşlarda başlaya ilerleyici serebellar ataksi ilk bulgudur. Başlangıçta gövdesel, ilerler ve zamanla ekstremite koordinasyonu bozulur Disartri, okulomotor apraksi görülebilir. Distoni ve koreatetozis hastaların yaklaşık %90’ında vardır ve yaş ilerledikçe belirgin hale gelir. 10 yaş civarında tekerlekli sandalyeye bağımlı hale gelirler. DTR azalmış veya alınamaz.

35
Ataksi Telenjektazi (AT) Telenjektaziler 2-4 yaş civadında ortaya çıkar. Kulak memesi, konjunktivalar, burun üstü, antekubital bölge ve dizde vardır. Serum IA, IgG ve IgE seviyeleri azalmıştır. Sık sinopulmoner enfeksiyon geçirirler. Zeka genellikle normaldir. Malignensi gelişme riski %38-40 civarındadır. (Lösemi, Hodgkin ve non-Hodgkin lenfoma) Serum AFP artmıştır. MRG; serebellar atrofi (erken dönemde normal olabilir) Destekleyici ve tekrarlayan enfesiyonlara yönelik tedavi uygulanır.
 Serum AFP artmıştır. MRG; serebellar atrofi (erken dönemde normal olabilir) Destekleyici ve tekrarlayan enfesiyonlara yönelik tedavi uygulanır..")
37
OR herediter ataksilerde erken, ayrıntılı fenotipik değerlendirme, laboratuar ve nörogörüntüleme çalışmalarının yapılması ayırıcı tanı açısından yardımcı olmaktadır. Ancak hastalıkların fenotipik heterojenitelerinin olması ve birbiri ile benzer özellikler içermeleri nedeni ile klinik olarak doğru tanı koymak olduça zordur. Bu nedenle klinik olarak şüphe duyulan hastalarda uygun mutasyon analizi istenmesi önerilmektedir.

Benzer bir sunumlar