Sunuyu indir
1
GENETİK DANIŞMA Doç. Dr. Zerrin Yılmaz Çelik

2
Bir hastalığın kalıtsal olduğu anlaşıldığında ya da şüphe edildiğinde, hastanın ailesi ve gelecekte aileye katılacak fertler için bu hastalığın tekrarlama riski gündeme gelir. Kalıtsal hastalıkların, aile bireyleri için riskini ve bu riskleri değiştirecek imkanları bildirmek genetik danışmanın ana konularından biridir. Riski değerlendirebilmek için genetik hastalığın tanısının konulmuş olması ve hastalık hakkında yeterli bilginin toplanması ön koşuldur.

3
Genetik bozukluk veya doğumsal anomalilere sahip bir aile üyesinin varlığında veya böyle bir bireye sahip olma riski olduğunda aileye hastalığın mevcut ve ortaya çıkabilecek sonuçları hakkında bilgi vermek aileyi olacaklara hazırlamak destek olmak genetik danışmanın amacıdır. Genetik danışma hastalığa yaklaşımın her aşamasında klinik takibin önemli bir parçası olmakla birlikte özel eğitim de gerektirmektedir.

4
Genetik danışma tıbbi genetik hizmetlerinin içinde yer alır
Genetik danışma tıbbi genetik hizmetlerinin içinde yer alır. Diğer uzmanlık dallarından farklı olarak tıbbi genetikte hastanın değerlendirilmesi; hasta ile ilgilenmenin yanı sıra uzun bir hazırlanmaya ve hastanın takibine gereksinim olması nedeniyle zaman alan bir uygulamadır. Genetik danışma, hastaya yaklaşımda genellikle ilk basamaktır. Danışma veren kişi tıbbi bilgisi yanında iyi iletişim kurma becerisine de sahip olmalıdır.

5
Genetik danışmanlık gerektiren durumlar
Kalıtsal bir hastalık için şüphe duyulması veya varlığının bilinmesi Doğum defektleri, multiple konjenital anomali Mental retardasyon İleri anne yaşı ve diğer nedenlerden dolayı doğum öncesi /imlantasyon öncesi genetik tanı gereken durumlar Tekrarlayan gebelik kayıpları Fertilite/ cinsel gelişim problemleri Akrabalık Genetik test yapılması/ genetik test sonucu anormallik+/-

6
GENETİK HASTALIKLAR Kromozom hastalıkları Tek gen hastalıkları
Multifaktöryel hastalıklar (poligenik) Atipik Kalıtım gösteren hastalıklar Sporadik veya ailesel kanserler
 Atipik Kalıtım gösteren hastalıklar. Sporadik veya ailesel kanserler.")
7
KROMOZOMAL HASTALIKLAR
SAYISAL BOZUKLUKLAR YAPISAL BOZUKLUKLAR SİTOGENETİK ve MOLEKÜLER SİTOGENETİK TANI YÖNTEMLERİ

8
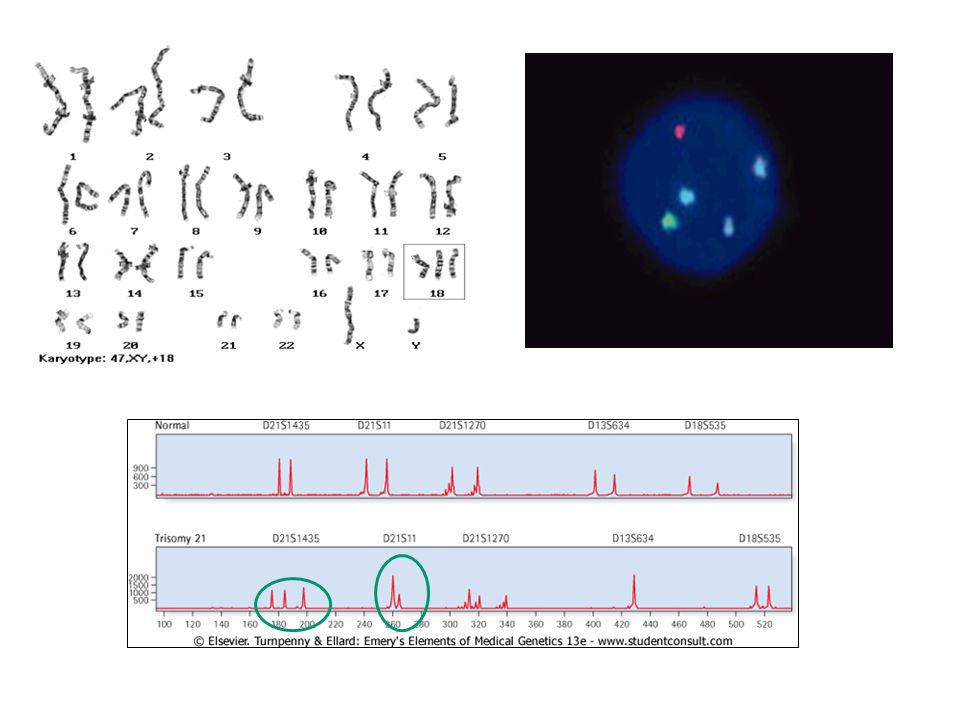
Sayısal kromozom anormallikleri
Anöploidiler en sık rastlanan anormalliktir. 13, 18, 21, cinsiyet kromozomu trizomileri X kromozomu monozomisi yaşamla bağdaşan anöploidilerdir. Bunlar dışındaki sayısal anormallikler gebeliğin erken dönemlerinde kaybedilir. İleri anne yaşı %95, baba yaşı %5 trizomik olgularda önem taşır. Cinsiyet kromozomu anöploidilerinde paternal mayotik hatalar önemlidir.

10
Spontan abortus olgularında kromozom anormallikleri
Trizomi %2 Trizomi %15 Trizomi %3 Trizomi %5 Diğer kromozom trizomileri %25 Monozomi-X %20 Triploidi %15 Tetraploidi %5 Diğer %10

11
Sayısal kromozom bozukluklarının tekrarlama riski diğer kromozom bozukluklarına göre düşük olmakla birlikte toplum riskinin üzerindedir. Ailede bir çocukta saptanırsa takip eden gebelikte prenatal genetik tanı önerilmelidir. Down sendromu gibi tipik sendrom bulguları klinik olarak tanınsa bile mutlaka sitogenetik tanı önerilmelidir. Gebelik takibinde ileri anne yaşının sayısal kromozom bozuklukları açısından risk oluşturduğu unutulmamalıdır.

12
Yapısal kromozom hastalıkları
Dengeli ve dengesiz olarak iki başlık altında toplanır. Dengeli yapısal kromozom bozuklukları, taşıyıcılar açısından risk oluşturmaz! Fenotip normaldir. (resiprokal translokasyon, insersiyon, robertson tipi translokasyon, inversiyon) Resiprokal translokasyonlar en sık bulgudur. Taşıyıcılarda dengesiz gamet oluşumu nedeniyle anomalili bebek doğumu, spontan abortus veya infertilite riski bulunur.
 Resiprokal translokasyonlar en sık bulgudur. Taşıyıcılarda dengesiz gamet oluşumu nedeniyle anomalili bebek doğumu, spontan abortus veya infertilite riski bulunur.")
13
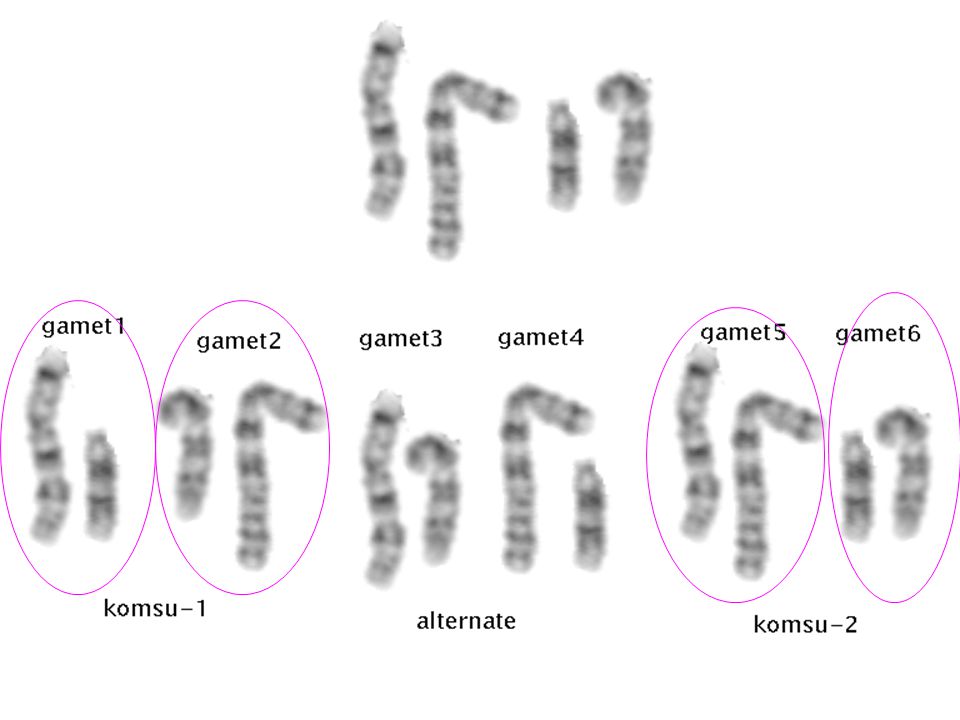
Komşu 2 Komşu 1 alternate

14
45,t(14;21)(q10;q10)
(q10;q10)")
15
Tekrarlayan gebelik kayıpları
İlk ve ikinci trimester gebelik kayıpları 3 veya daha fazla ise eşlerden sitogenetik analiz istenmelidir Eşlerden birinde dengeli translokasyon olasılığı %3-5 dir. Eşlerden birinde dengeli translokasyon saptanırsa bu ailenin yeni gebeliğinde prenatal tanı yapılmalıdır. İnfertilite sorunu olan böyle bireylere preimplantasyon genetik tanı seçeneği sunulmalıdır.

16
Anomali tipi Taşıyıcı Amniyosentezle anomali riski rob(13;14) Her iki eş %1 rob(14;21) Baba Anne %15 rob(21;22) %5 %10 rob(21;21) %100 Resiprokal translokasyon %12 İnversiyon %50
 %5. %10. rob(21;21) %100. Resiprokal translokasyon. %12. İnversiyon. %50.")
17
Dengesiz yapısal bozukluklar
Mental retardasyon ve multiple konjenital anomaliler dengesiz yapısal kromozom bozukluklarına eşlik eder. Delesyon en sık bulgudur, duplikasyon ve marker kromozom bunu takip eder. Bireyde dengesiz yapısal anomali varlığı anne babasında kromozom incelemesi yapılmasını gerektirir. Turner S. da X kromozomunda delesyon izokromozom ve ring oluşumu görülebilir.

18
Kromozom bozukluklarının sonuçları
Mental retardasyon Multiple konjenital anomali Kromozomal sendromlar Cinsel gelişim kusurları Spontan abortus Tekrarlayan gebelik kaybı İntrauterin ex Neonatal ölüm Ölü doğum

19
Cinsiyet kromozomlarında bozukluk? Fertilite problemleri
Gecikmiş puberte Adet görememe Erken menopoz Cinsiyet farklanmasında bozukluk Çocuk sahibi olamama

20
Sitogenetik tanı yöntemleri
Genetik hastalık riski durumunda tanı ve danışmanlık için önem taşır. Kromozom hastalıklarının tanısını koyar. Trizomilerde, regüler veya translokasyon tipi bozukluğun ayırıcı tanısını koyar. Hastalığa neden olan kromozom bozukluğunun sayısal ya da yapısal olduğunun bilinmesi sonraki gebelikte var olan riski belirlemede gereklidir. Olguda mozaikliğin var olup olmaması klinik seyir açısından önem taşır. Sendromik bireylerde tesadüfen saptanan yapısal kromozom bozuklukları klinik bulguya neden olan gen bölgesini gösterebilir.

21
TEK GEN BOZUKLUKLARI Hastalığa tek bir gen içindeki mutasyon neden olur. *Kalıtım genin üzerinde bulunduğu kromozoma ve etkinliğine bağlı olarak adlandırılır: Otozomal a. Dominant b. Resesif X’e bağlı a. Dominant

22
*Nükleer kromozomlar kadar mitekondriyel kromozomlar üzerindeki genlerde de mutasyon olabilmektedir.
Mitokondriyel hastalıklar genellikle sinir ve kaslarda enerji üretimini etkiler ve hücresel yaşlanmada rol oynar. Tek gen hastalıkları yenidoğan ve erken çocukluk dönemlerinde belirgindir.

23
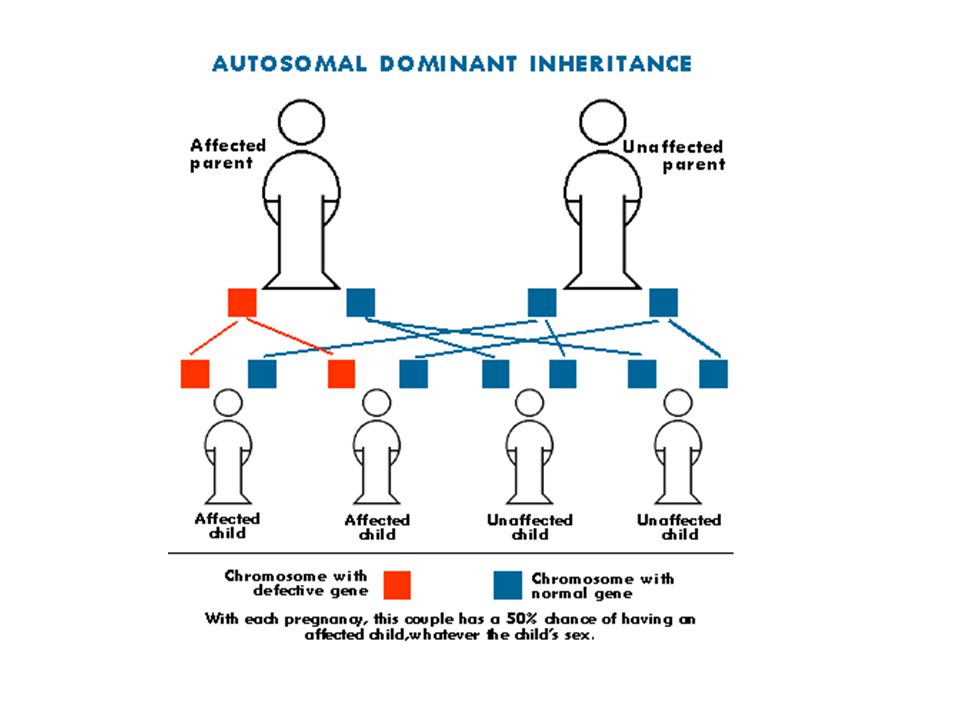
Otozomal dominant hastalıklar
Hasta bireylerin çocuklarına %50 olasılıkla hastalık aktarılır Birey yeni mutant ise kardeşleri açısından risk yoktur. Genetik danışma açısından sorunlu durumlar Penetrans eksikliği Ekspressivite değişkenliği Gonadal mozaisizm Pedigri analizi ve aile incelemesi detaylı yapılmalıdır. Hastalığın penetransı ve klinik bulguları iyi bilinmelidir.

25
2 3 1 Aa Aa Aa Aa Aa Aa Aa Aa Aa

26
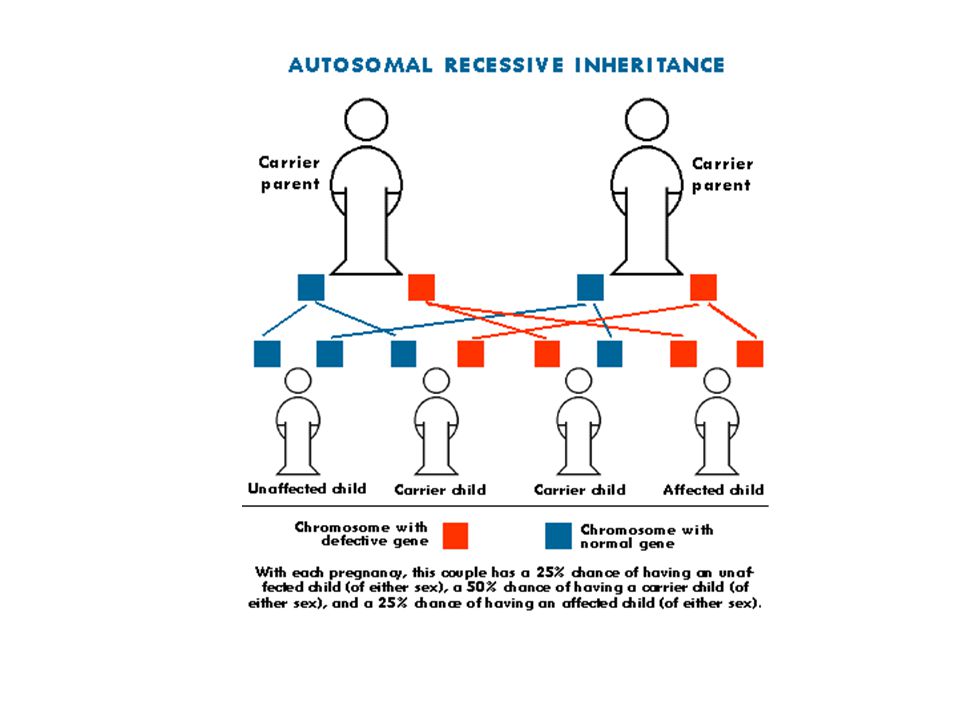
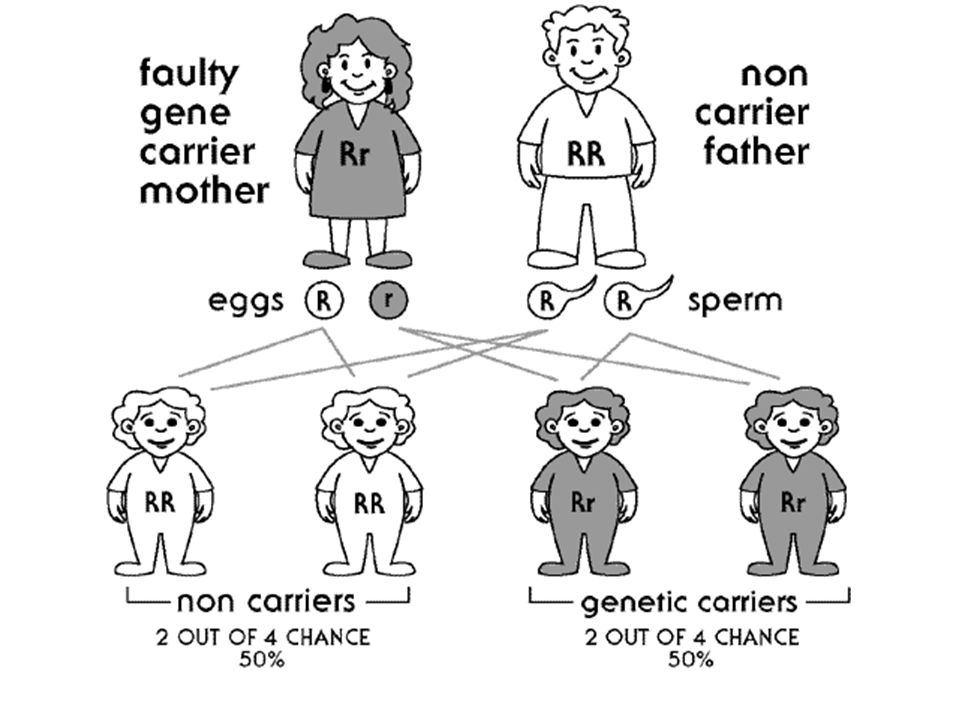
Otozomal resesif hastalıklar
En sık rastlanan otazomal gen hastalıklarıdır. Genellikle hasta çocuğu olan aileler başvurur. Bu bireyler hastalık için zorunlu taşıyıcıdır ve doğacak çocuklar açısından hastalık riski ¼ dür. Akraba evlilikleri bu grup hastalıklar açısından riski arttırmaktadır ancak, her bireyin birkaç tane nadir, zararlı resesif mutant gen Taşıdığı unutulmamalıdır. Taşıyıcılar belirlenmelidir. Allelik heterojenite varlığında birleşik heterozigotluk sonucu hastalık gelişebilir.

30
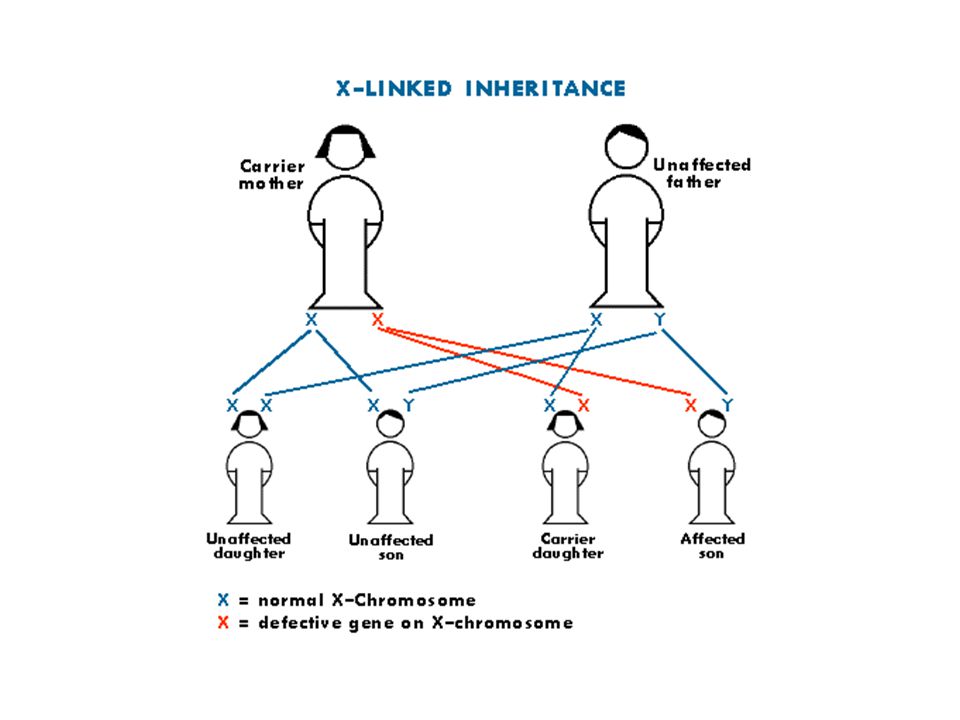
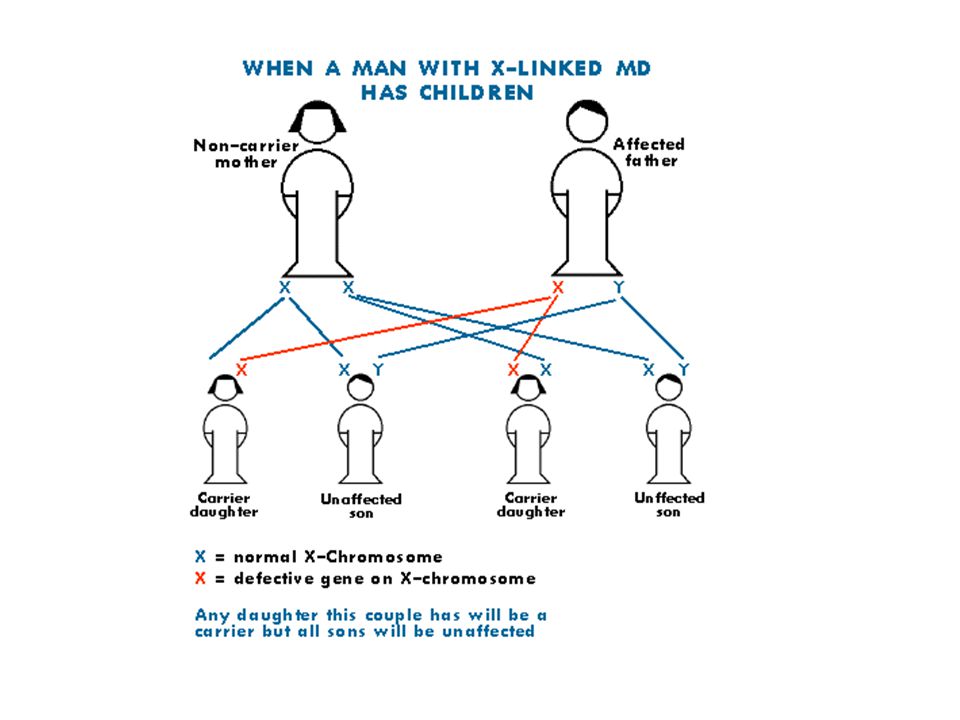
X’e bağlı resesif hastalıklar
Hemen daima erkeklerde görülür. Ancak babadan oğluna geçmez. Kadınlar fenotipik normal taşıyıcıdır. X inaktivasyonu sonucu hastalık genini taşıyan X aktif kalırsa kadınlarda da hastalık bulguları görülebilir. Taşıyıcıların belirlenmesi genetik riskler açısından önemlidir. Hasta bireyin anne ve babası sağlıklı ise (de novo mutasyon) sonraki gebelikte hastalık riski yoktur.
 sonraki gebelikte hastalık riski yoktur.")
33
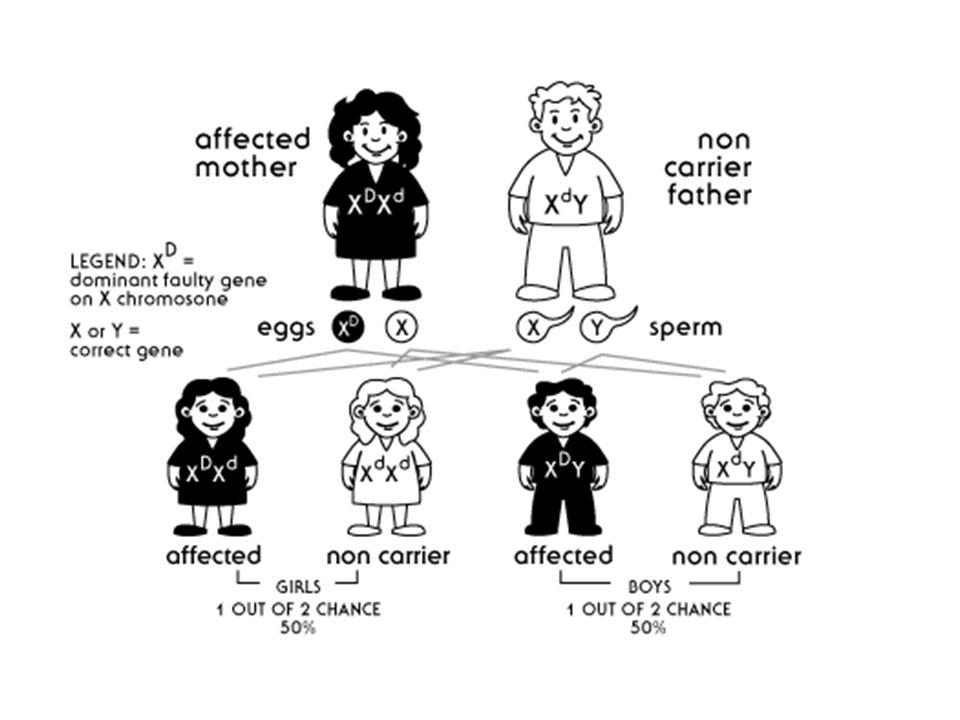
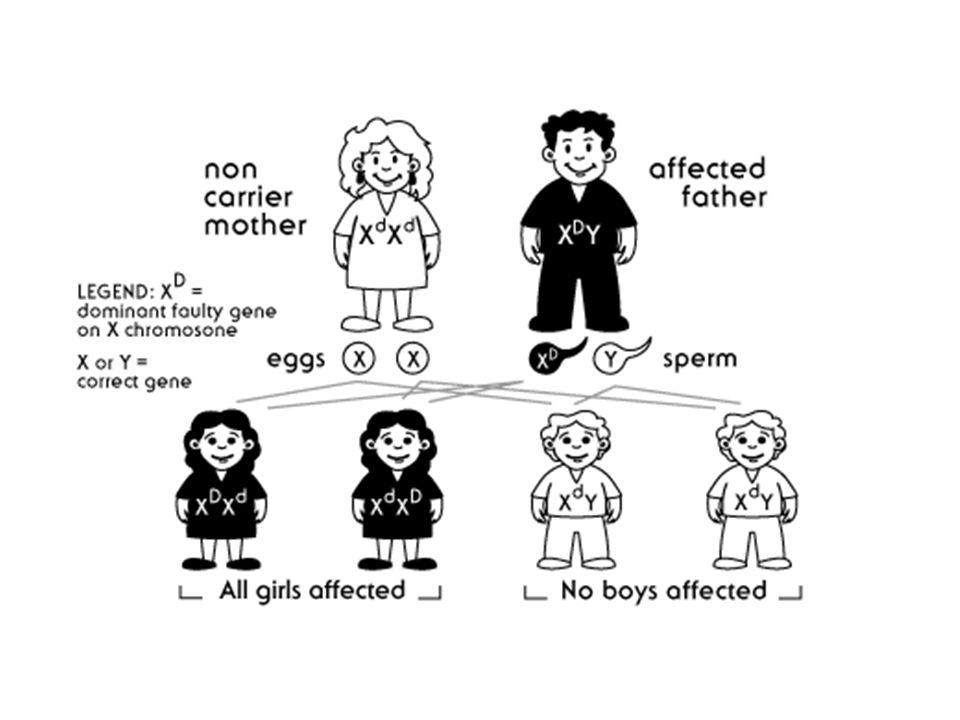
X’ e bağlı Dominanat kalıtım
Kalıtım tipi pedigride otozomal dominanat hastalıklar gibidir. Tek fark hastalığın babadan oğuluna geçmemesidir. Bu tip hastalıklar genellikle erkekler için öldürücüdür. Hastalık dişi bireylerde tanımlanır.

36
Mitokondriyel kalıtım
Hastalık anneden aktarılır, hasta babanın çocuklarında risk yoktur. Nükleer genlerdeki mutasyonlara bağlı olarak da mitokondri fonksiyonları bozulabilir, bu durumda kalıtım maternal değildir. Klinik heterojenite heteroplazmi ile açıklanabilir.

37
Moleküler genetik tanı yöntemleri
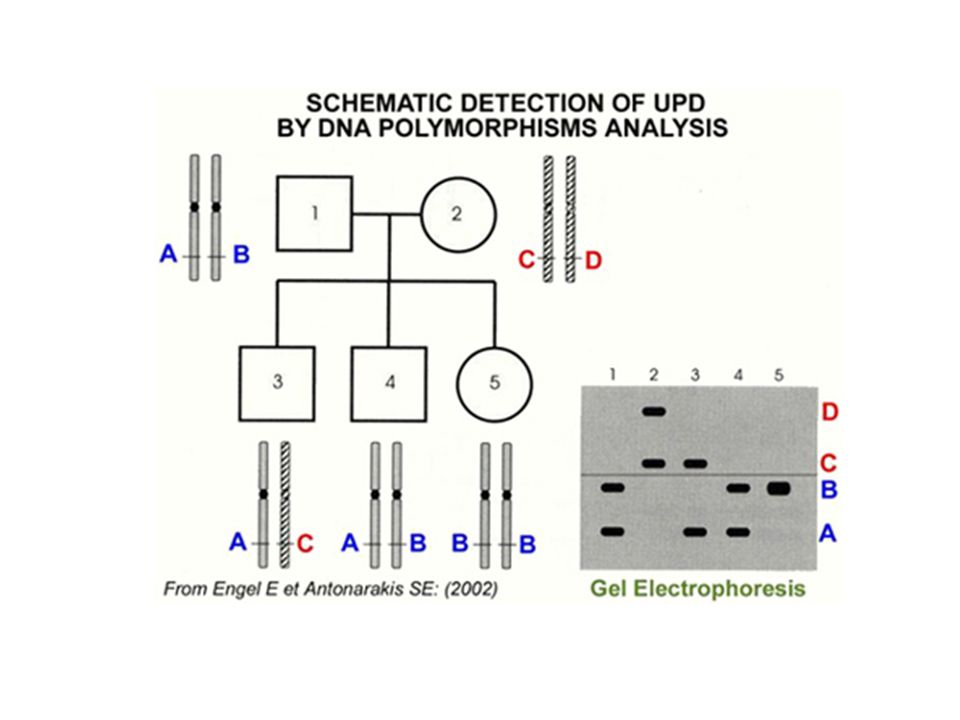
Gen bozukluğu bilinen mendeliyan hastalık açısından riskli durumlarda postnatal ve prenatal tanı imkanı sağlar. (PCR-RFLP analizleri) Mutasyonu belirlenememiş olgularda işaretleyiciler kullanarak bağlantı analizi ile informatif aile bireyi varlığında hastalığın tanısı koyulabilir ( PCR – aile çalışması- haplotipleme). STR markerları ile parental analize olanak sağlayarak sitogenetik anormallik görülmeyen uniparental dizominin tanısını koyar. Tanı ve ayırıcı tanı yapılmasını sağlayarak genetik danışmanın içeriğini belirler.
 Mutasyonu belirlenememiş olgularda işaretleyiciler kullanarak. bağlantı analizi ile informatif aile bireyi varlığında hastalığın. tanısı koyulabilir ( PCR – aile çalışması- haplotipleme). STR markerları ile parental analize olanak sağlayarak sitogenetik. anormallik görülmeyen uniparental dizominin tanısını koyar. Tanı ve ayırıcı tanı yapılmasını sağlayarak genetik danışmanın. içeriğini belirler.")
38
Alfa2 globin ekzon dizi analizi

40
SRY

41
Multifaktöryel (poligenik) hastalıklar
Bu hastalıklar minör genler tarafından oluşturulur. Çevre etkisi hastalığın ortaya çıkmasında önem taşır. Yetişkinlerde görülen hipertansiyon, diyabet,şizofreni kadar çocuklarda görülen yarık damak, yarık dudak, NTD ve konjenital kalp hastalığı gibi değişikliklere neden olur. Kronik hastalıklara yol açtıkları için yetişkinlerde daha yaygın görülmekle birlikte çocuk mortalitesinin %25-35’ inden sorumludurlar. Bu hastalıkların bir ailede bulunması , o aile bireyleri açısından riskin, toplumun genel riskinden kat fazla olmasına yol açar.

42
Ailede tekrarlama riskini arttıran durumlar
İki hasta çocuktan sonra Toplumda seyrek bir hastalıksa Hastalık propozitusda ağır seyrediyorsa Propozitus o hastalık için az görülen cinsiyette ise Propozitusun yakın akrabalarından biri hasta ise (ailede 1’den fazla kişi hasta ise)
")
43
Akraba evliliklerinde genetik danışma
Bu çiftler için Anomalili bebek sahibi olma riski toplum riskinin iki katıdır %4.5-5 2. Resesif hastalık taşıyıcılığı açısından hasta bebek sahibi olma olasılıkları rasgele evliliklerden yüksektir 3. Taşıyıcı sıklığını arttırırlar Çocuk sahibi olduklarında diğer gebelik 3-4 yıl sonra düşünülmeli (geç bulgu veren hastalıklar açısından)
")
44
Hastalık açısından risklerin değişmesine neden olarak
genetik danışmada problem oluşturan durumlar vardır Probandın tanısının olmaması Yanlış veya eksik tanı Genetik heterojenite Penetrans eksikliği Ekspressivite çesitliliği Gonadal mozaisim Dengesiz mutasyonlar

45
Genetik danışmada vakaya yaklaşım
Bilgi toplama aile hikayesi, pedigri analizi tıbbi hikaye yapılan test sonuçlarının değerlendirilmesi Fizik muayene ve gerekli testlerin yapılması Bilgilendirme hastalığı nedenleri ve klinik seyri tekrar riskleri ileri tetkik veya ileride uygulanabilecek testler karar verme gerekli konsultasyonların yapılması Takip aile ile iletişimin devamlı olması hastalığın takibi ve olası risk değişimlerinin gerekirse tekrar konuşulması açısından önem taşır. Geç bulgu veren hastalıklar veya tanımlanamayan durumlarda klinik takip önemlidir.

46
Danışmanlık Genetik danışma oturumları sırasında aileye söz konusu
genetik hastalık hakkında uygun ve yeterli bilgi aktarılmalıdır. Ailelerin genellikle bilmek istedikleri hastalığın tekrarlama riskidir. Aileye tüm olasılıklar ve bu olasılıklara ilişkin tüm seçenekler anlatılmalıdır. Tekrarlama risklerini önlemek için seçeneklerin ne olduğu anlatılmalı.

47
Karar Aile bilgilendirildikten sonra çocuk yapıp yapmama, gebeliğin
terminasyonu veya tanı testi yaptırıp yaptırmama gibi konularda karar vermelidir. Gerekirse aileye bu karar için süre verilmelidir. Genetik danışman karar verme konusunda yönlendirme yapmamalıdır.

48
Genetik hastalıklar için tanı yöntemler
Hastalık tipi incelenen materyal tanısal testler Kromozom hastalıkları Kromozom *Pedigri analizi Sitogenetik ve moleküler sitogenetik inceleme Tek gen hastalıkları DNA Moleküler genetik yöntemler Multifaktöryel hastalıklar Sitogenetik ve moleküler genetik yöntemler Mitokondriyal kalıtım hastalıkları Mitokondriyal DNA Moleküler yöntemler *Tüm hastalıklar için ilk uygulanan işlemdir

49
Genetik danışmanın aşamaları
Olası risk tanımlanır. Neden test yapılıyor, İleri anne yaşı Kullanılacak yöntem anlatılır. Prenatal genetik tanı yöntemleri İşlemin yapılabilmesi için bilgilendirilmiş onam formu doldurulmalıdır (Bu form endikasyonlar, işlemin uygulama ve sonuçları hakkında bilgi içermelidir.) Sitogenetik tanı için bilgilendirilmiş onam formu Sonuçlar çıkınca ikinci oturumda sonucun ne olduğu anlatılır ve neler yapılacağı planlanır. Down S. , normal karyotip veya polimorfik kromozom saptanabilir
 Sitogenetik tanı için bilgilendirilmiş onam formu. Sonuçlar çıkınca ikinci oturumda sonucun ne olduğu anlatılır ve neler yapılacağı planlanır. Down S. , normal karyotip veya polimorfik kromozom saptanabilir.")
50
Multiple konjenital anomali Metabolik hastalık ?
Mental Motor Retardasyon Sitogenetik inceleme *Periferik kandan kromozom eldesi *GTG bantlama (550band düzeyi) *Analiz
 *Analiz.")
51
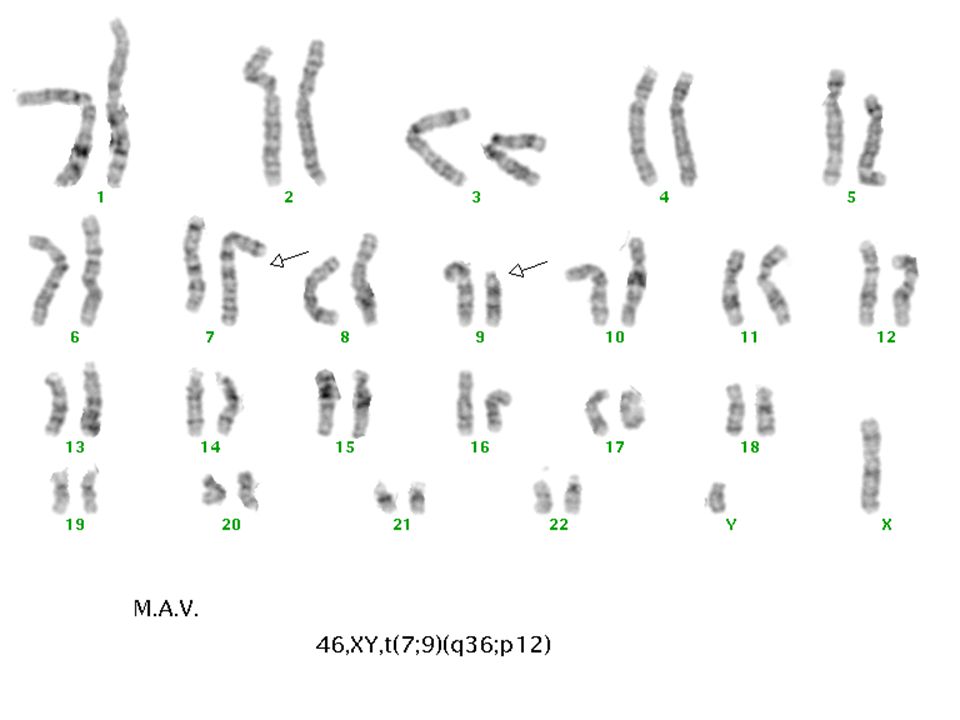
46,XY,7q+(?)
")
52
Aile ile konuşarak randevu verildi
Oturum Pedigri çıkarıldı Karyotipte saptanan bulgu hakkında bilgi verildi Anne- Baba karyotipinin yapılması önerildi

56
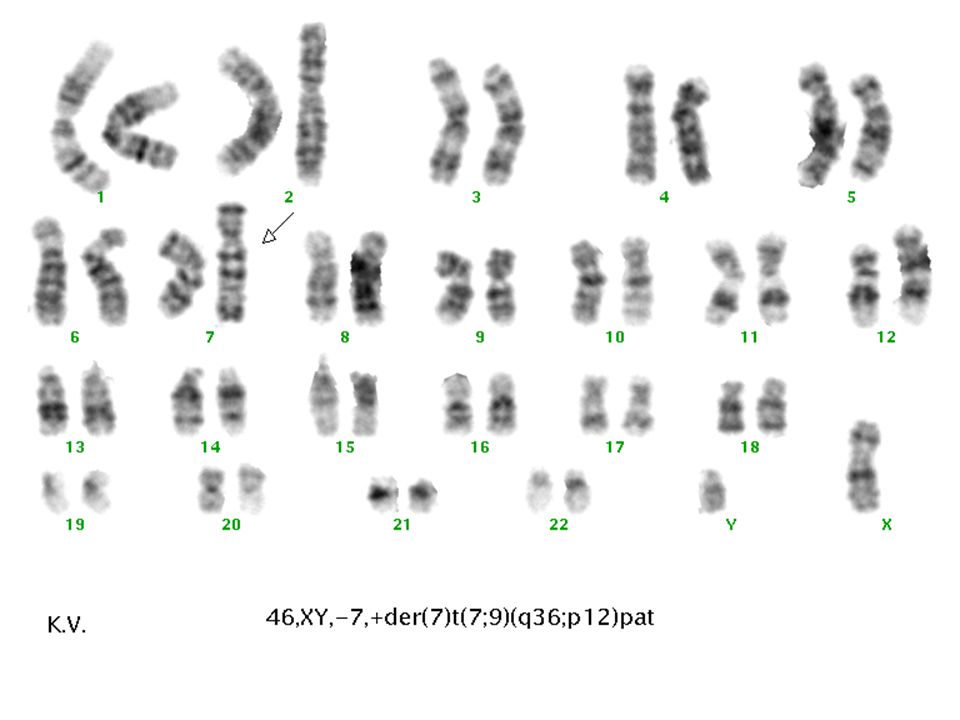
2. Oturum Aileye saptanan bulgular ve bunun riski anlattıldı Çocuktaki durum tekrar açıklandı Ailenin sağlıklı ilk çocuklarına ve Babanın anne ve babasına karyotipleme yapılması önerildi

58
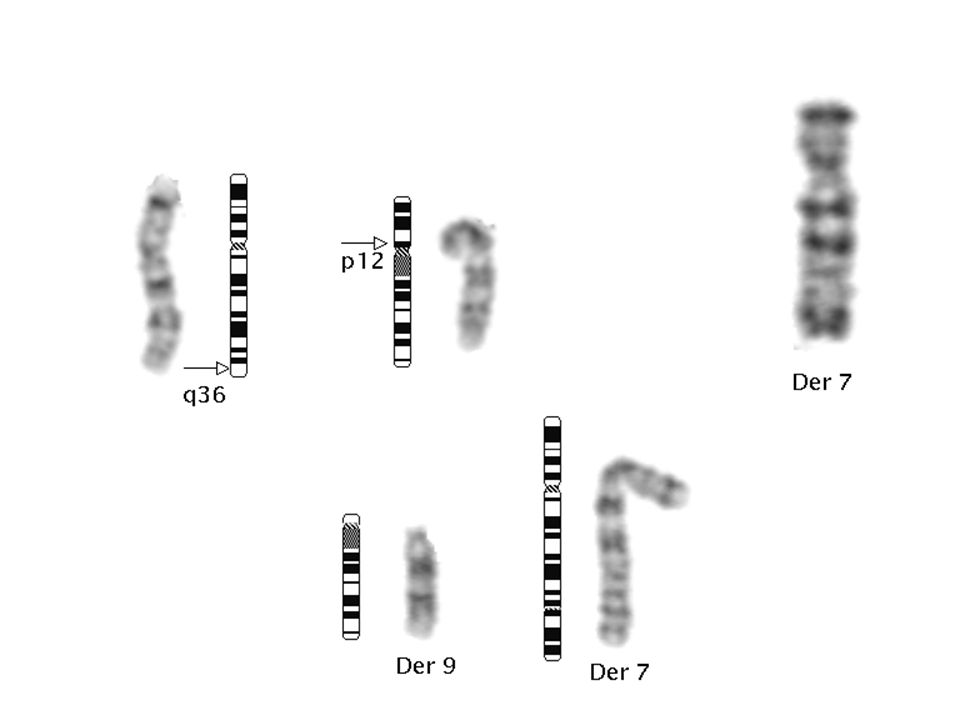
Parsiyel trizomi 9p (Rethoré sendromu)
Psikomotor retardasyon Mikrosefali Konjenital kalp defekti Hipertelorizm Böbrek anomalileri Downslanting palpebral fissür Kalça ve diz eklemi çıkıkları Geniş burun Kranial stur anomalileri Kısa üst dudak Aşağı doğru kavisli ağız köşeleri Hafif iri malforme kulak Kifoskolyoz Küçük el ve ayaklar Karakteristik dermatoglifik özellikler Klinodaktili, Simian Çizgisi Kriptoorşitizm

59
Monozomi 7q36 Holoprosensefali Siklopi Ciddi santral sinir sistemi malformasyonları

60
Genetik danışma ne işe yarar?
Ailenin karşılaştığı riski anlaması. Bu konuda yapılacak tıbbi girişimler konusunda bilgilenerek karar vermesi. Var olan genetik bozukluğu anlaması ve buna yönelik sorularına cevap alması. Gebe kalma, gebeliğini devam ettirip ettirmeme, evlat edinme kararını verebilmesi. Sonraki gebelikler için plan yapması. Endişelerinin giderilmesini sağlar.

61
Genetik danışmada bazı yanlış inanışlar düzeltilmelidir:
-Doğumda bulunan bütün hastalıklar ve malformasyonların etyolojisinin genetik temele dayalı olduğu düşüncesi. -Kromozomal hastalıkların devamlı şekilde kalıtsal olduğu. -Tüm genetik hastalıkların kromozom analizi ile ortaya çıktığı. -Hasta kişinin benzer durumda bir kardeşi yoksa bu hastalığın genetik olduğu konusunda tereddütlerin olması. -Otozomal dominant hastalıklarda hastalığın yalnızca bayanlarda olduğu. -Otozomal resesif bir hastalık açısından tasıyıcı olan anne babadan hasta çocuk doğma riski %25 ise, hasta bir çocuktan sonra bu riskin sonraki doğacak üç çocuk için ortadan kalktığı düşüncesi. -Genetik tetkikler sonrasında mutlaka sonuç alınacağı.



>")


















