Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Mitokondrial DNA hastalıklarına PGD ile tanı konulabilir mi. Prof. Dr
Mitokondrial DNA hastalıklarına PGD ile tanı konulabilir mi? Prof. Dr. Muhterem BAHÇE GENLAB Genetik Hastalıklar Tanı Merkezi ANKARA

2
http://bhavanajagat. wordpress
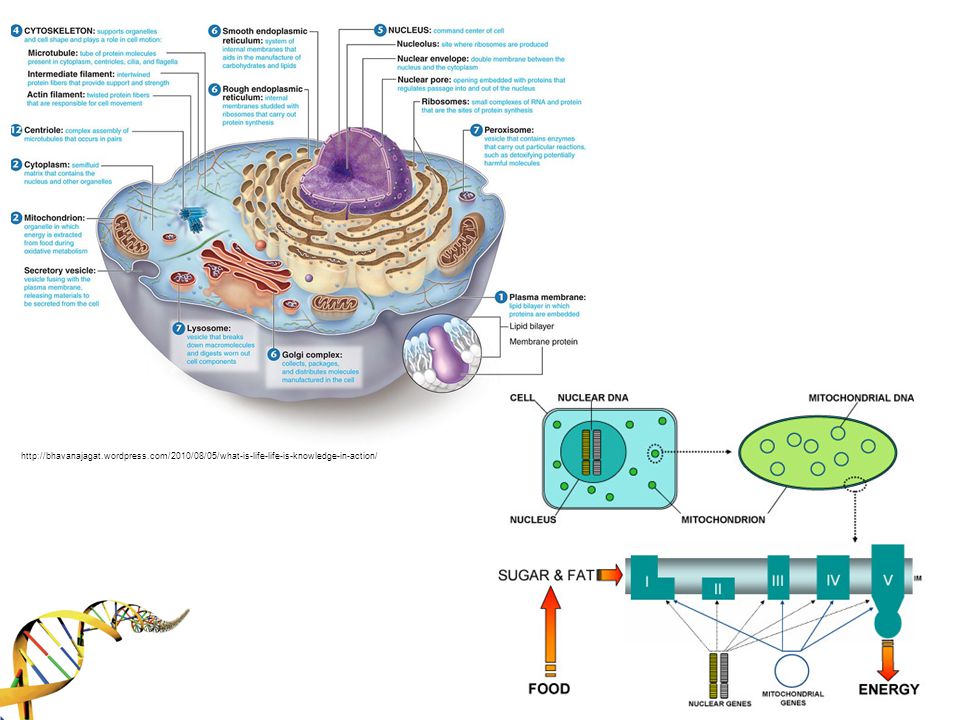
3
*Mitokondrinin kökeni primitif bir aerobik bakteriye (α-proteabacteria) dayanmaktadır. *Gelişim sürecinde ökaryot içine girmiştir (endosimbiyoz) *MtDNA’nın moleküler markerlarının takibi ile ilk kadının yıl önce Afrika’da yaşadığı sonucuna varılmıştır.
 *MtDNA’nın moleküler markerlarının takibi ile ilk kadının yıl önce Afrika’da yaşadığı sonucuna varılmıştır..")
4
Elektron transport zinciri için gerekli 13 gen içerir (67 gen nükleer DNA )
16,569 baz çift uzunluğunda sirküler bir DNA’ya sahiptir.

5
Mitokondri sayısı Oosit. : 100 000 Blastosist. : 1 000
Mitokondri sayısı Oosit : Blastosist : Primordiyal germ hücresi :

6
Mitokondri fonksiyonları için yaklaşık 1500 nükleer gene gereksinim vardır. Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PPARGC1A) - Bu gen mutasyonları da hastalıklara neden olabilir. 150’den fazla patolojik mtDNA mutasyonu tanımlanmıştır. - 1/5000 doğumda bir mtDNA’ya bağlı hastalık görülmektedir. Bu oran fetüslerde daha yüksektir.
 - Bu gen mutasyonları da hastalıklara neden olabilir. 150’den fazla patolojik mtDNA mutasyonu tanımlanmıştır. - 1/5000 doğumda bir mtDNA’ya bağlı hastalık görülmektedir. Bu oran fetüslerde daha yüksektir..")
7
Mitokondrial DNA (mtDNA) hastalıkları
NARP(Neurogenic muscle weakness, Ataxia, Retinis Pigmentosa)/Leigh MELAS (Mitochondrial myopathy, Encephalopathy, Lactic acidosis, and Stroke-like episodes) LHON (Leber Hereditary Optic Neuropathy) Özel mtDNA mutasyonları
/Leigh. MELAS (Mitochondrial myopathy, Encephalopathy, Lactic acidosis, and Stroke-like episodes) LHON (Leber Hereditary Optic Neuropathy) Özel mtDNA mutasyonları.")
8
Mitokondriyal hastalıklardan etkilenen sistemler ve semptomlar
Kalp Kardiyomiyopati Kalp bloku Pre eksitasyon sendromu Renal Renal tübüler defektler Endokrin Hipoparatiroidizm Hipotiroidizm Gonadal yetmezlik Sindirim Disfaji Konstipasyon Karaciğer yetmezliği Periferik sinir sistemi Miyopati Aksonal sensorimotor nöropati Santral sinir sistemi Ensefalopati Stroke-benzeri nöbetler Epilepsi ve bunama Psikozis ve depresyon Ataksi Migren Göz Eksternal oftalmopleji Ptozis Katarakt Pigmenter retinopati Optik atrofi İşitme Bilateral sensorinöral sağırlık

10
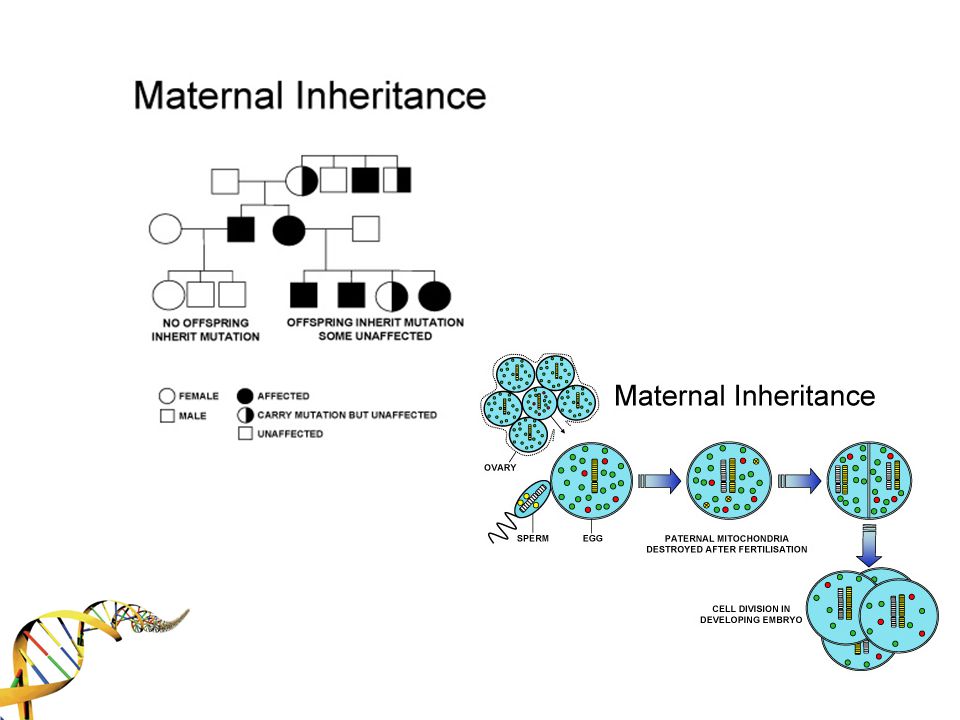
HOMOPLAZMİ HETEROPLAZMİ

12
MELAS (m. 3243A>G)(MT-TL1) NARP/Leigh Synd. (m
MELAS (m.3243A>G)(MT-TL1) NARP/Leigh Synd. (m.8993T>G)(MT-ATP6) 32 embriyo ve 12 fetüste MtDNA’nın dağılım şekilleri incelenmiş. m.3243A>G’nin dokularda ve oogenezde rasgele m.8993T>G ise oogenezde daha belirgin, dokularda rasgele
(MT-TL1) NARP/Leigh Synd. (m.8993T>G)(MT-ATP6) 32 embriyo ve 12 fetüste MtDNA’nın dağılım şekilleri incelenmiş. m.3243A>G’nin dokularda ve oogenezde rasgele m.8993T>G ise oogenezde daha belirgin, dokularda rasgele.")
13
MtDNA segregasyonu *Mutant MtDNA beklenenden daha az oranda sonraki kuşağa aktarılıyor. Negatif seleksiyon için farklı teoriler geliştirilmiş: 1. MtDNA hasarına bağlı hastalığı olan bireyler reprodüktif çağa ulaşmadan hayatlarını kaybediyorlar Follikülogenez ve oogenezde mtDNA sayısı minimal düzeye indiriliyor ve fenotipi olumsuz etkilenen mitokondriler mitofaj ile elimine ediliyor. Daha sağlıklı olanlar sonraki kuşağa aktarılıyor. 3. Erken oogenez aşamasında Golgi etrafında endoplazmik retikulum ve mitokondrilerden oluşan “Balbiani cisimcikleri” görülüyor. Bu yapıya seçilmiş mitokondriler giriyor ve sonraki nesile bu yolla aktarım oluyor.

14
Mitokondrial hastalıkların Prenatal veya Preimplantasyon Genetik Tanısı nDNA mtDNA PND PGD PND PGD

15
Prenatal tanı CVS AZ mutant yük ÇOK mutant yük ARA düzey
Prenatal tanı CVS AZ mutant yük ÇOK mutant yük ARA düzey ??? Farklı dokularda farklı dağılım??? Aynı dokuda zaman içinde mutasyon yükünün değişebilir??? Mutant doz ve hastalık ilişkisi ??? NARP, (neurogenic weakness, ataxia and retinitis pigmentosa ) mtDNA 8993T>G veya T>C
 mtDNA 8993T>G veya T>C.")
16
PGD 1. PB 2. Blastomer 3. Blastosist

17
MELAS. : m. 3243A>G mutasyonu (MT-TL1) (n = 30), MERRF. : m
MELAS : m.3243A>G mutasyonu (MT-TL1) (n = 30), MERRF : m.8344A>G mutasyonu (MT-TK) (n = 15), LEIGH : m.9185T>G mutasyonu (MT-ATP6) (n = 6) PB materyalinin analizleri sonucunda mtDNA mutasyon (heteroplazmik) analizi için bu yöntemin güvenilir olmadığı gösterilmiştir.
 (n = 30), MERRF : m.8344A>G mutasyonu (MT-TK) (n = 15), LEIGH : m.9185T>G mutasyonu (MT-ATP6) (n = 6) PB materyalinin analizleri sonucunda mtDNA mutasyon (heteroplazmik) analizi için bu yöntemin güvenilir olmadığı gösterilmiştir.")
18
PGT’nin güvenilirliği 1
PGT’nin güvenilirliği 1. Mutasyon yükü ve hastalığın klinik ağırlığı arasındaki korelasyona 2. Mutant mtDNA’nın tüm blastomerlerde eşit oranda dağıldığı varsayımına 3. Mutant yükün zamanla (prenatal ve postnatal) değişmeyeceği varsayımına bağlıdır.
 değişmeyeceği varsayımına bağlıdır.")
19
NARP ve Leigh sendromu (m. 8993T>G ve m
NARP ve Leigh sendromu (m.8993T>G ve m.8993T>C) bu kriterlere uymaktadır. Ancak her bir olgu tek tek değerlendirilmelidir. MELAS(m.3243A>G) doku dağılımı değişkendir ve zamanla mutasyon yükü değişebilir. %30 mutant yük sonrası bulgu verebilir.
 bu kriterlere uymaktadır. Ancak her bir olgu tek tek değerlendirilmelidir. MELAS(m.3243A>G) doku dağılımı değişkendir ve zamanla mutasyon yükü değişebilir. %30 mutant yük sonrası bulgu verebilir.")
20
In spite of the benefits provided by PND and PGD strategies, they are unable to provide an absolute guarantee that offspring will not develop symptoms of the disorder and their validity is extremely limited in cases where mtDNA mutation load is poorly correlated with disease severity.

21
8. Conclusions We conclude that genetic management of mtDNA diseases is improving and that PGD of mtDNA disease holds a great deal of promise, offering diagnostic advantages over traditional pre-natal methods of testing. Although therapeutic interventions for children and adults affected by mtDNA disorders remain extremely limited, there have been significant advances in the preconception correction of mtDNA disease (i.e. in oocytes), via cytoplasmic and nuclear transfer on experimental basis. However, at this time, such radical approaches to manage transmission of mutant mtDNA are forbidden in most countries. Before such methods can be considered as a viable approach to human disease, much additional work in model organisms, proving safety, efficacy, and improving efficiency, is required.
, via cytoplasmic and nuclear transfer on experimental basis. However, at this time, such radical approaches to manage transmission of mutant mtDNA are forbidden in most countries. Before such methods can be considered as a viable approach to human disease, much additional work in model organisms, proving safety, efficacy, and improving efficiency, is required.")
22
Teknik olarak mitokondri transferi veya nükleus transferi ile mtDNA mutasyonuna bağlı hastalıkların sonraki kuşağa geçişi önlenebilmektedir. Tartışılması gereken konu üç ebeveynli bir bireyin durumu ve bu işlemlerin hangi şartlar altında uygulanacağıdır.

Benzer bir sunumlar









.>")




 DİPLOMA EKİ EĞİTİM SEMİNERİ 2011-2013 Dönemi Bologna Sürecinin Türkiye’de.>")


>")



