Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
ZİHİNSEL ENGELLİ ÇOCUKLARIN FİZİKSEL GÖRÜNÜMLERİ
MEHMET ŞİMŞEK Zih. Eng. Öğretmeni

2
Zihinsel Engellilik: Gelişimsel dönemde ortaya çıkan, uyumlu davranışlarda görülen yetersizliğe ilaveten genel zeka fonksiyonları açısından normalin altında olma durumudur. Zihinsel engelli bireyleri tanılamada birçok test ve yöntem kullanılmaktadır. Zihinsel yetersizliği olan bireyi tanılamada yardımcı olabilecek en önemli etkenlerden bir tanesi onların fiziksel görünüm özellikleridir. Bu özellikleri sahip oldukları tanıya göre açıklayabiliriz.

3
MİKROSEFALİ: HİDROSEFALİ POLYDACTLY SYNDACTLY DOWN SENDROMU FRAJİL X SENDROMU CORNELLA DE LANGE SENDROMU WİLLİAMS SENDROMU RUBİNSTEİN TAYBİ SENDROMU TÜBEROSKLEROZ STURGE-WEBER SENDROMU DANDY-WALKER SENDROMU TURNER SENDROMU

4
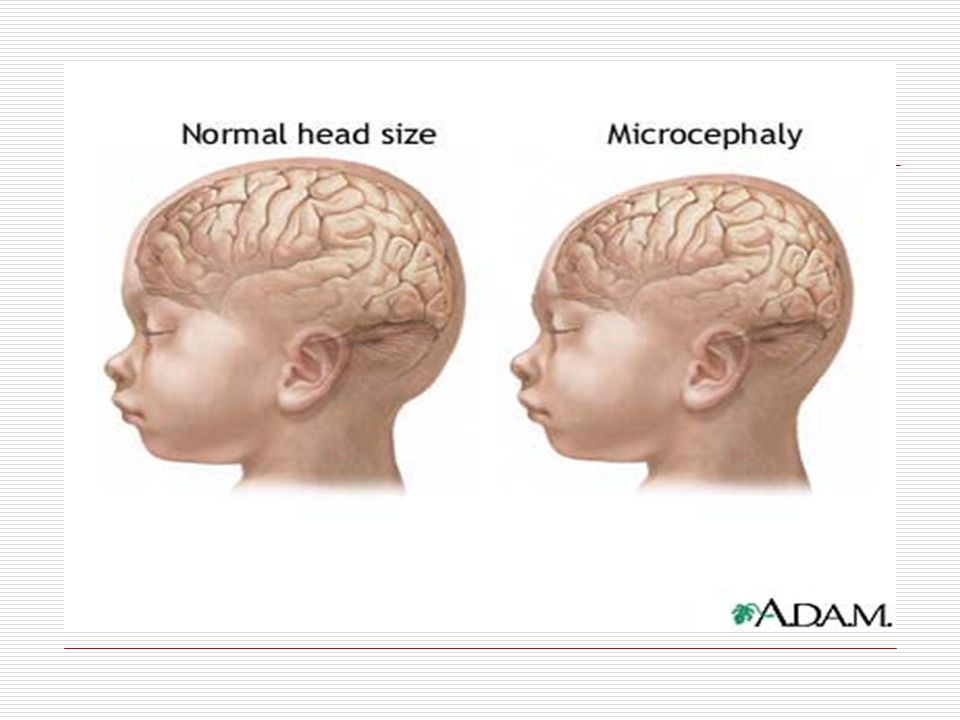
I. MİKROSEFALİ: Mikrosefali bir hastalık olarak tanımlanmasından daha çok klinik bir bulgudur. Mikrosefali; yaş ve cinsiyet ortalamasının üç standart sapmadan daha küçük ölçülen baş çevresi olarak tanımlanır. Küçük kafa yapısına sahiptirler ve ağır zihinsel engellidirler. Normal gelişim gösteren bir çocukta doğumda baş çevresi ortalama 35cm, üç aylık bir bebekte 40,5 cm altı aylıkta 43cm, bir yaşında yaklaşık 46cm dir. Bu çocuklarda ise; Kafa ufak, alın eğiktir. Kafa yapıların çoğunlukla yuvarlaktır. Yürürken vücut pozisyonu sarkık ve öne doğrudur. Mikrosefali 2 gruba ayrılır

5
A) Birincil Mikrosefali: Gebeliğin ilk 7 ayında olan gelişimin sonucunda ortaya çıkar. Genetik, kromozomal bozukluklar (Down sendromu, Edward sendromu, Cornelia de lange sendromu, rubinstein taybi sendromu), radyasyon, doğumsal enfeksiyonlar, kimyasal ilaçlar nedenleri ile ortaya çıkabilmektedir. Croniostosis (kafatası kemikleri arasındaki sütürlerin erken kapanıp kemikleşmesi) ten sonra teşhis edilir. B) İkincil Mikrosefali: Gebeliğin son iki ayında yada parinatal dönemde ortaya çıkar.
 İkincil Mikrosefali: Gebeliğin son iki ayında yada parinatal dönemde ortaya çıkar.")
9
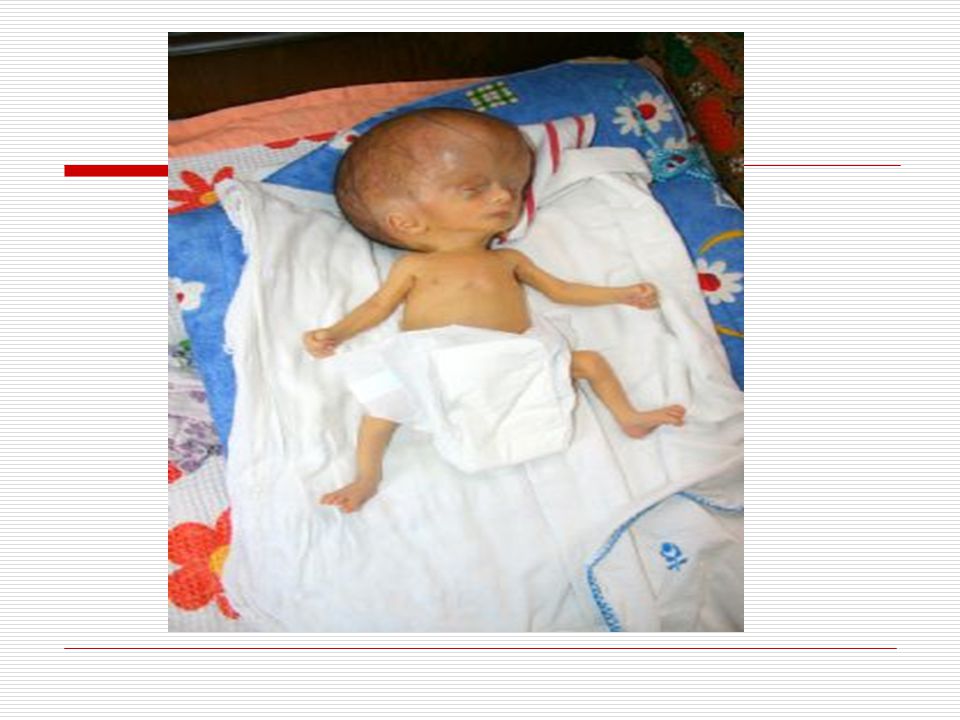
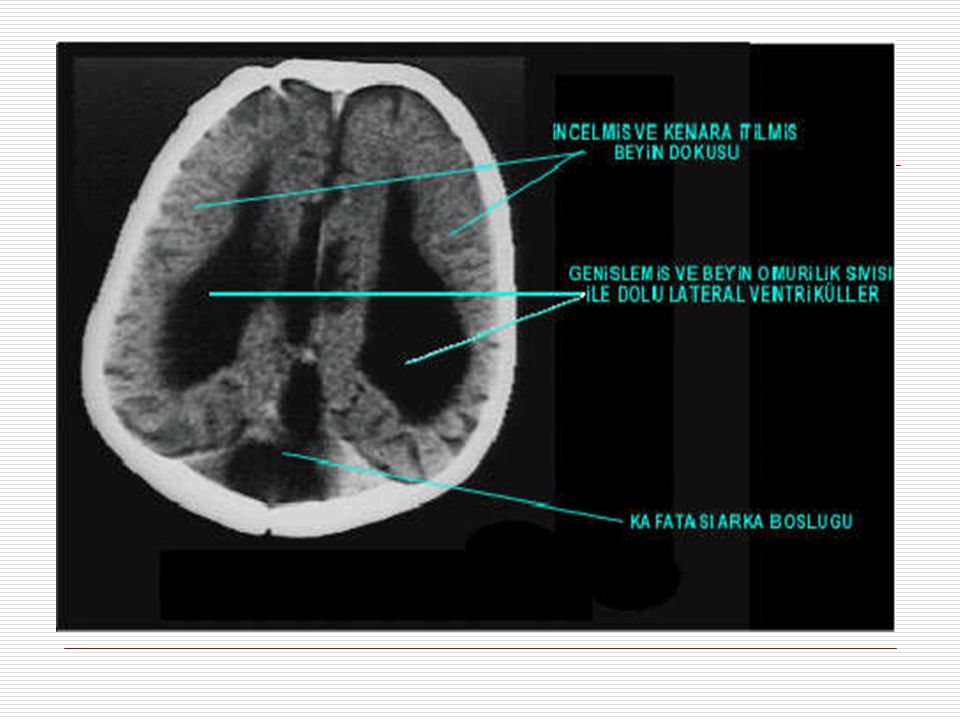
II. HİDROSEFALİ: Kafatası boşluğunu dolduran ve volumun %10 unu BOS denilen bir sıvı doldurur. Beynin korunması, ihtiyaçlarının ve atıklarının taşınmasını sağlayan bu sıvının üretimi ve emilimi arasındaki dengesizlik kafa içinde basıncın, artmasına yani hidrosefaliye sebep olur. Hidrosefalide bebek ve küçük çocuklarda kafatası kemikleri tam kapanmadığı için kafatası basıncın etkisi ile genişler. Doğumda çocuk normal olarak görülebilir ama yaşamın ilk yıllarında baş büyümesi belli olur. Bazı belirgin özellikleri; Kafa; normal gelişim gösteren çocuklara oranla büyüktür. Art kafa çıkıntılıdır. Alın; çıkık, düz ve küçüktür (başın öteki kısımlarına göre). Aynı zamanda daha ön tarafta ve belirgindir. Göz akı, renkli tabakanın üstünde belirgin bir şekilde görülür. Optik diskler genellikle soluktur.
. Aynı zamanda daha ön tarafta ve belirgindir. Göz akı, renkli tabakanın üstünde belirgin bir şekilde görülür. Optik diskler genellikle soluktur.")
13
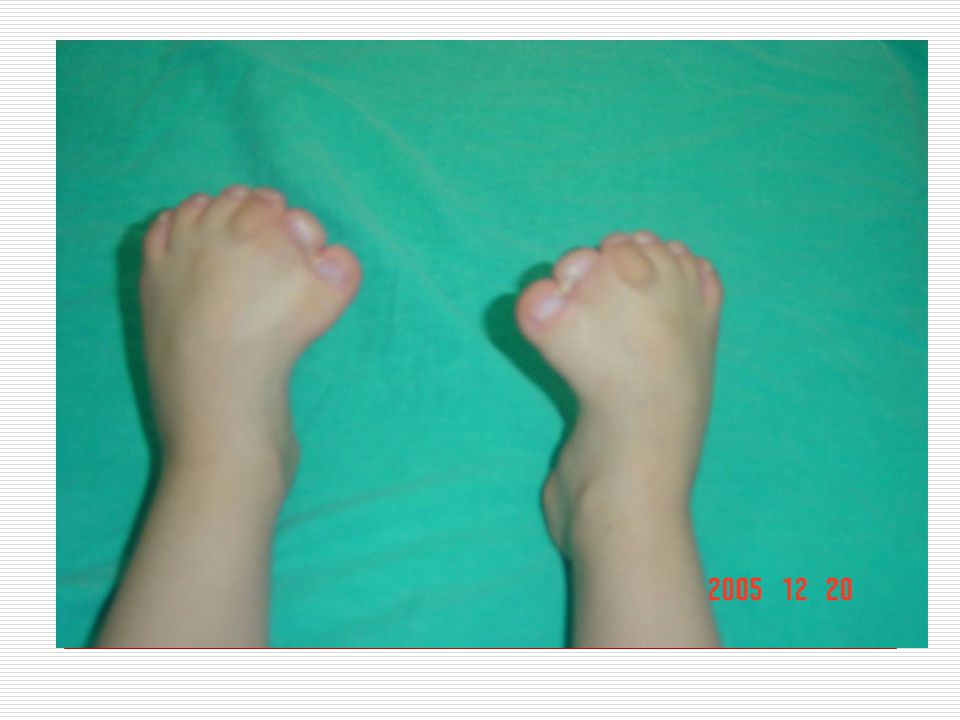
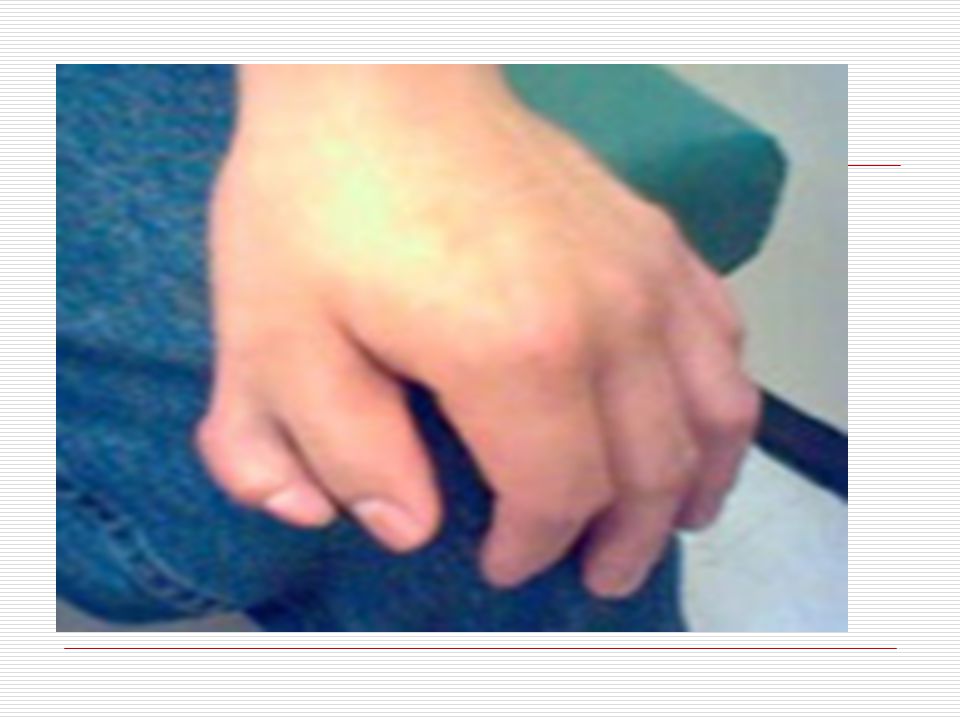
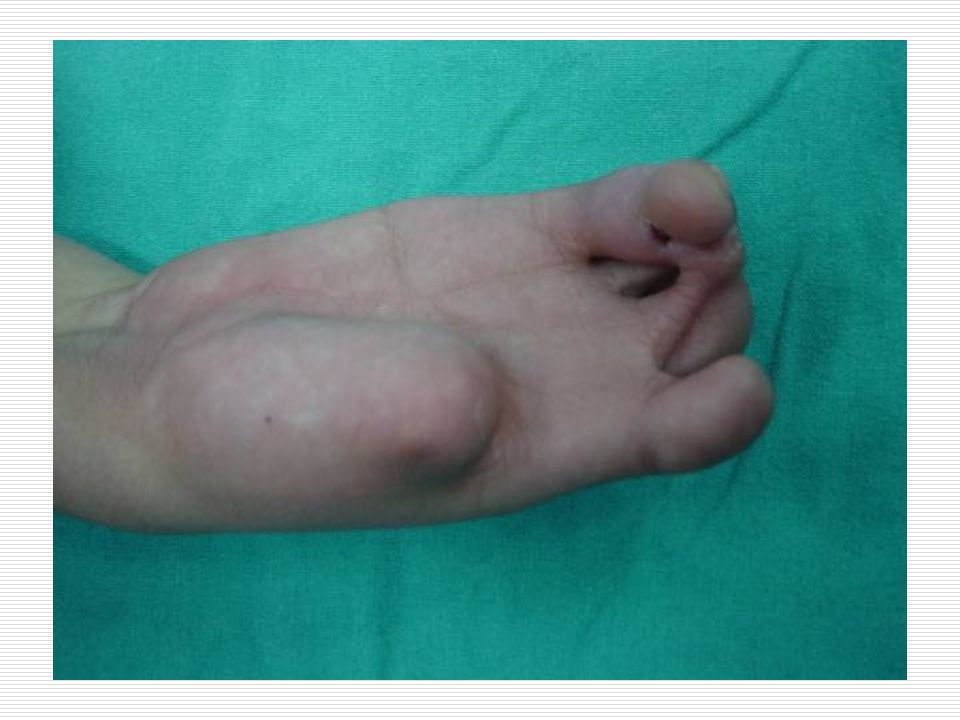
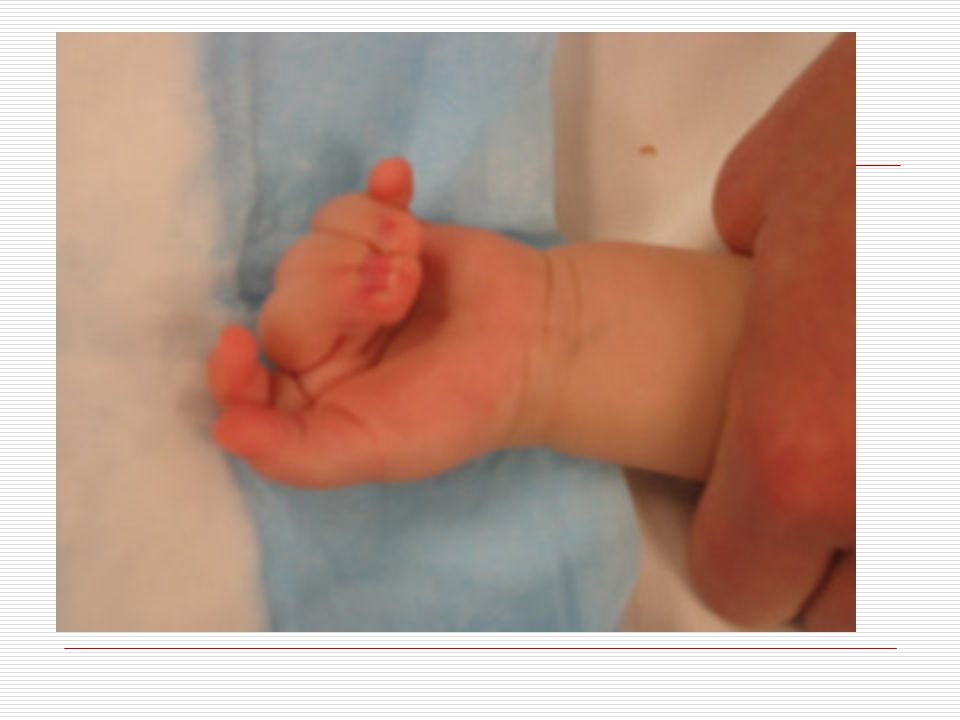
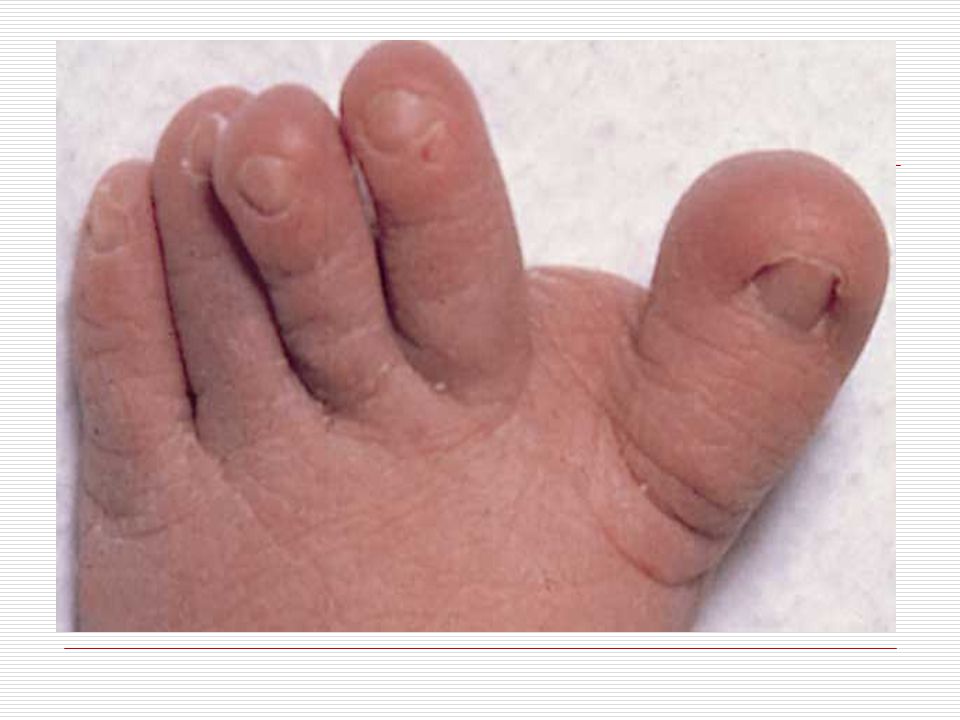
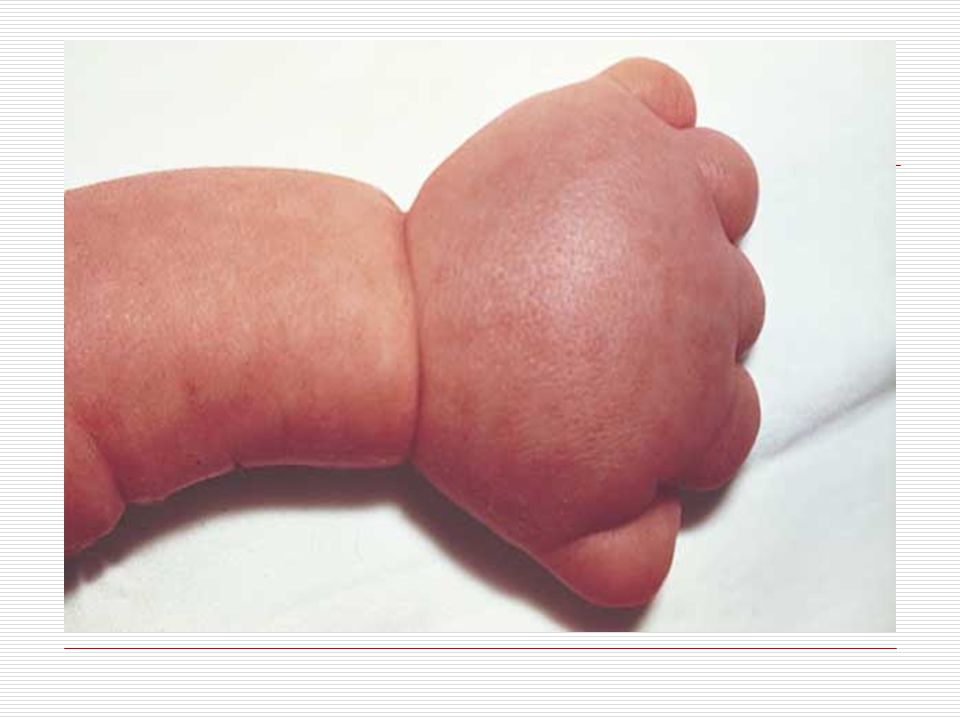
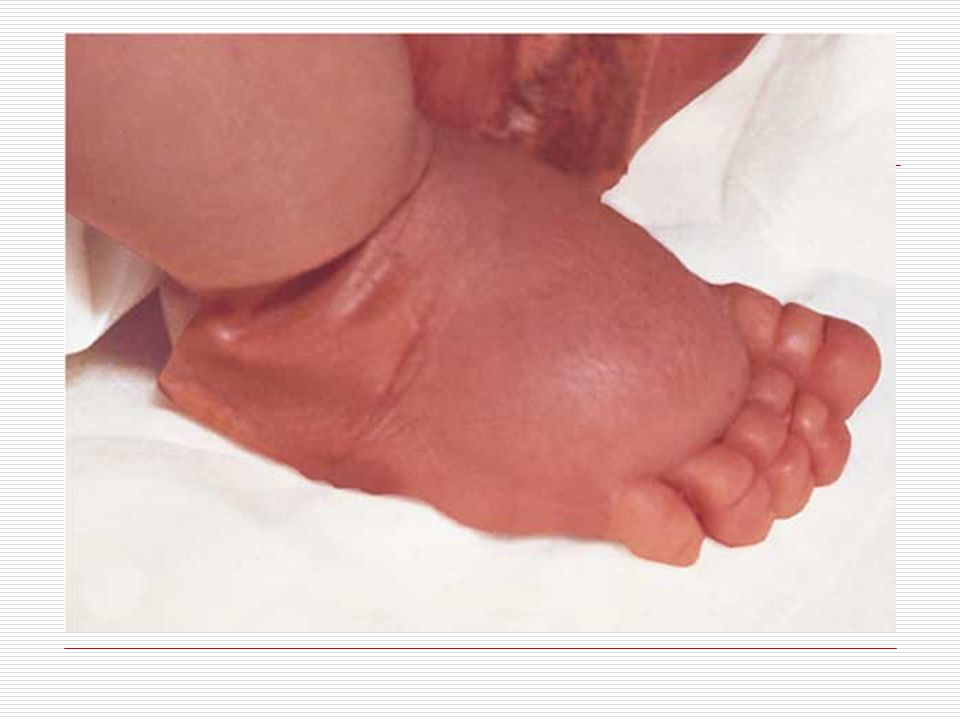
III. POLİDAKTİLİ: El veya ayak parmaklarında normalde olması gerekenden daha fazla sayıda doğuştan parmak bulunması durumudur. Anne karnında el ve ayak yapıları gelişirken parmaklar önce yapışık durumdadır. Gelişim sırasında parmak aralarındaki dokuların erimesiyle parmaklar ayrılır. Ancak bazen parmakların ayrılması sırasında bir hata sonucu fazla sayıda parmaklar ya da polidaktili oluşabilir. Ortalama 500 doğumda bir görülen polidaktili bazen sindaktili ile birliktede görülebilmektedir. Polidaktili farklı şekillerde olabilir. Başparmak ya da küçük parmağın yanında ufak bir şişlik gibi olabilir, bir parmağın uç kısmında iki uç şeklinde çatallanma gibi olabilir, elin kenarında hiç hareket etmeyen ufak bir sapla bağlı bir parmak şeklinde olabilir ya da tam gelişmiş hareketli normal bir fazla parmak şeklinde olabilir. Polidaktili ameliyatla düzeltilebilir. Ancak ameliyat sonrasında az yada çok iz kalacaktır.

19
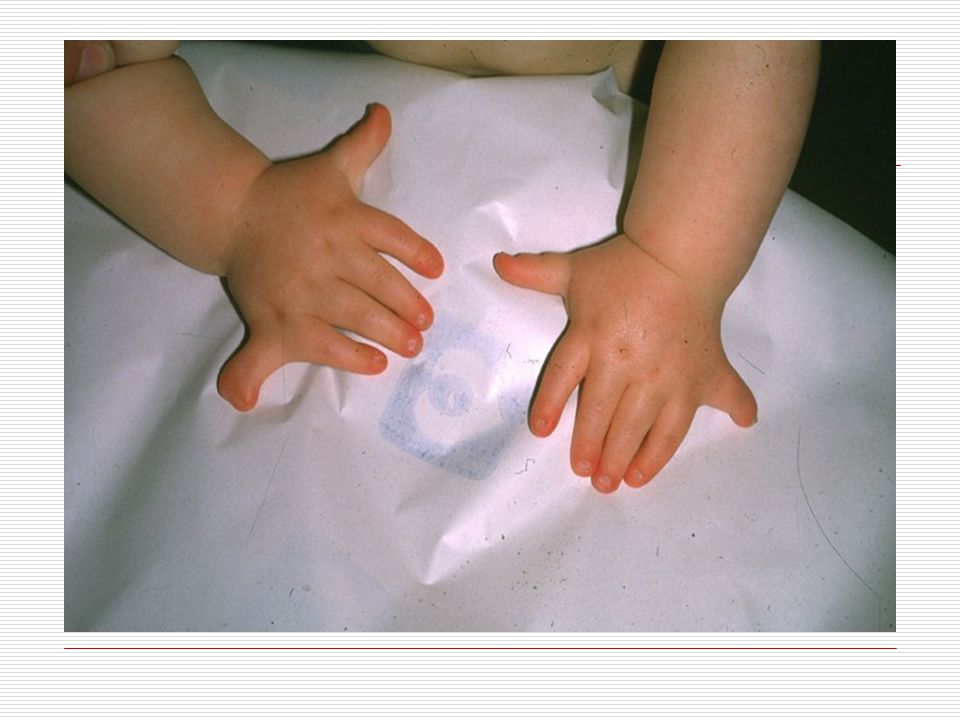
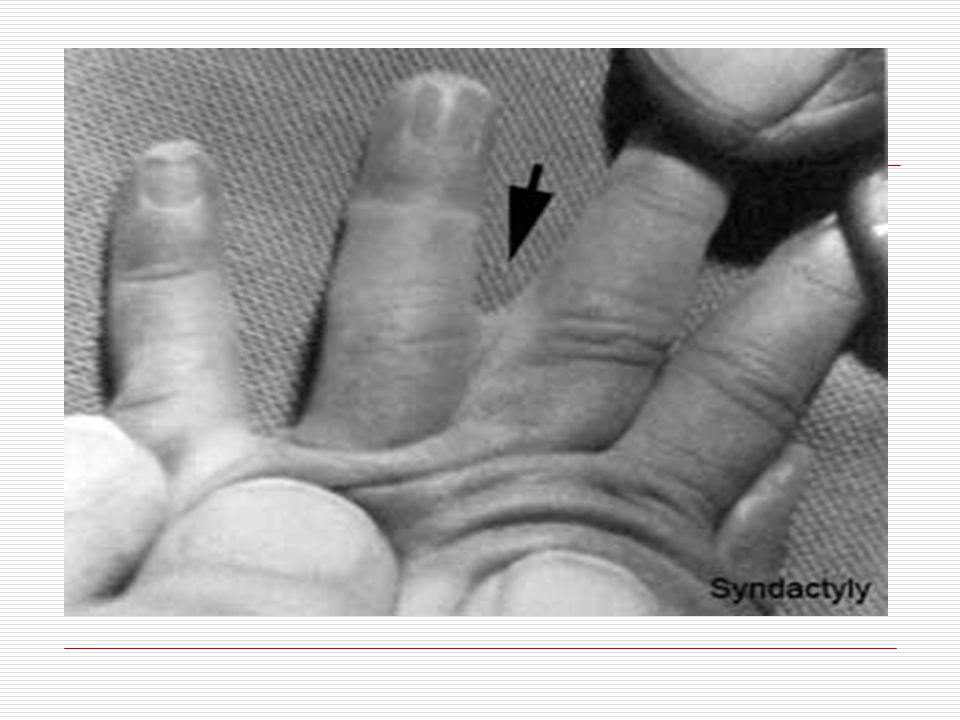
IV. SİNDAKTİLİ: Doğuştan el ya da ayak parmaklarının yapışık olması durumuna "sindaktili" denir. Görülme sıklığı 2000 canlı doğumda birdir. Hastaların % 50 sinde bir tarafta ( sağ veya sol) görülür. Erkeklerde daha sıktır. Kalıtımla da geçme olasılığı vardır. En fazla 3. ve 4. parmakların yapışıklığı görülür. Sindaktili 3 farklı tipte görülür. Bunlar; a) Komplet: İskelet sistemi normaldir. Tüm parmak birbirine yapışıktır. b) Inkomplet: İskelet sistem normaldir. Parmağın proksimal (ana iskelete yakın olan) yarısında yapışıklık mevcuttur. c) Kompleks: Iskelet sistemde de füzyon mevcuttur. Sindaktili; Apert sendromu, Poland sendromu, Gordon sendromunda çounlukla görülür. Bunun dışında Dady- Waker, Rbeinstein Taybi, Cornella de lange ve Down sendromunda da sıklıkla görülebilmektedir.
 görülür. Erkeklerde daha sıktır. Kalıtımla da geçme olasılığı vardır. En fazla 3. ve 4. parmakların yapışıklığı görülür. Sindaktili 3 farklı tipte görülür. Bunlar; a) Komplet: İskelet sistemi normaldir. Tüm parmak birbirine yapışıktır. b) Inkomplet: İskelet sistem normaldir. Parmağın proksimal (ana iskelete yakın olan) yarısında yapışıklık mevcuttur. c) Kompleks: Iskelet sistemde de füzyon mevcuttur. Sindaktili; Apert sendromu, Poland sendromu, Gordon sendromunda çounlukla görülür. Bunun dışında Dady- Waker, Rbeinstein Taybi, Cornella de lange ve Down sendromunda da sıklıkla görülebilmektedir.")
24
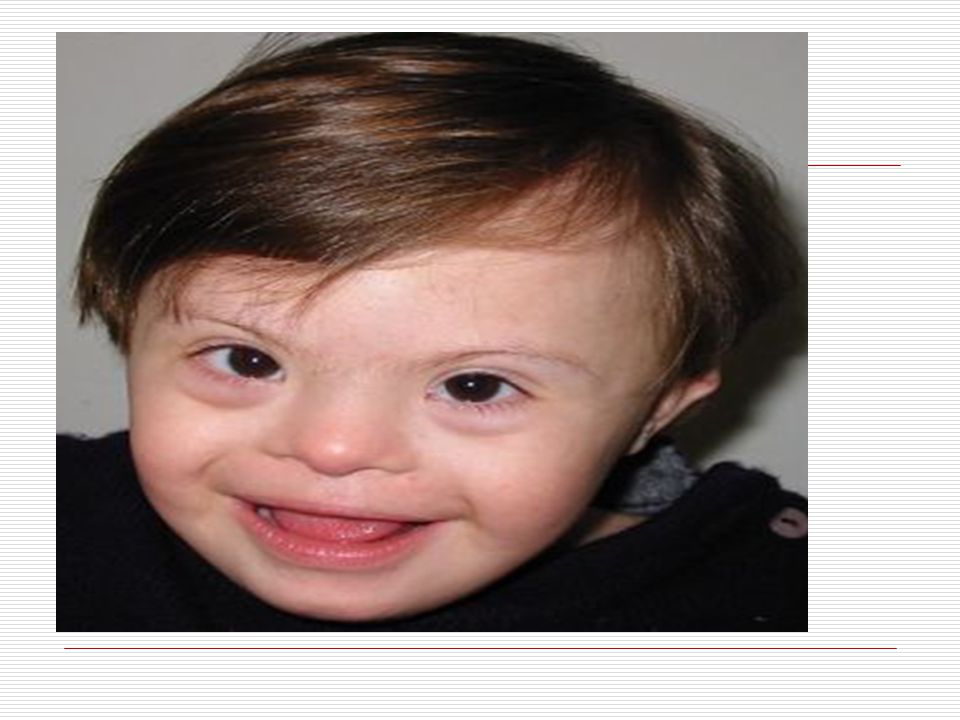
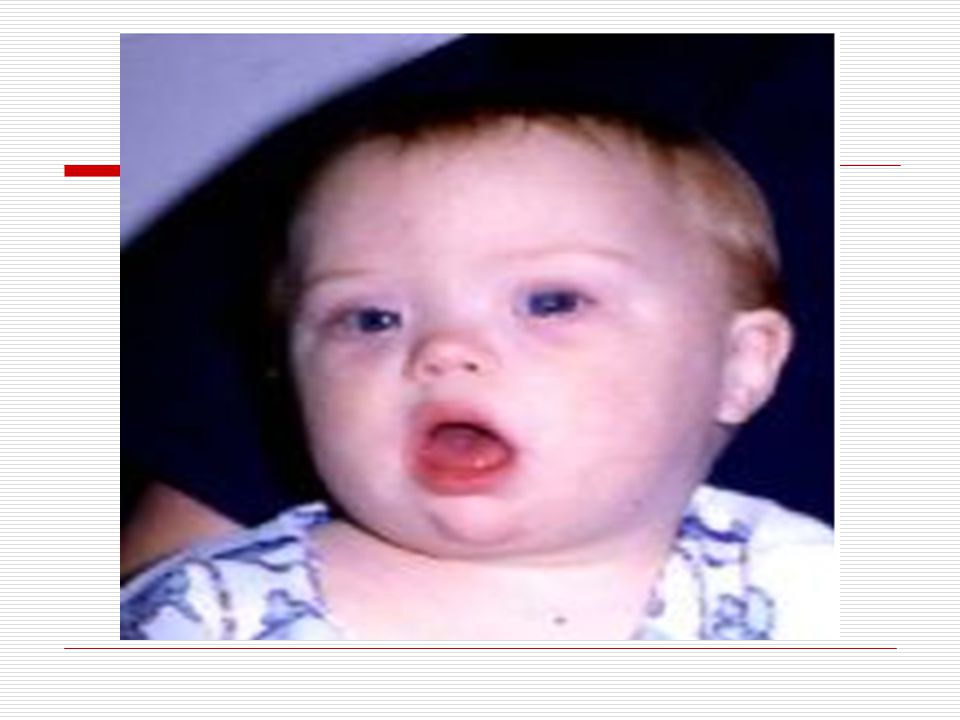
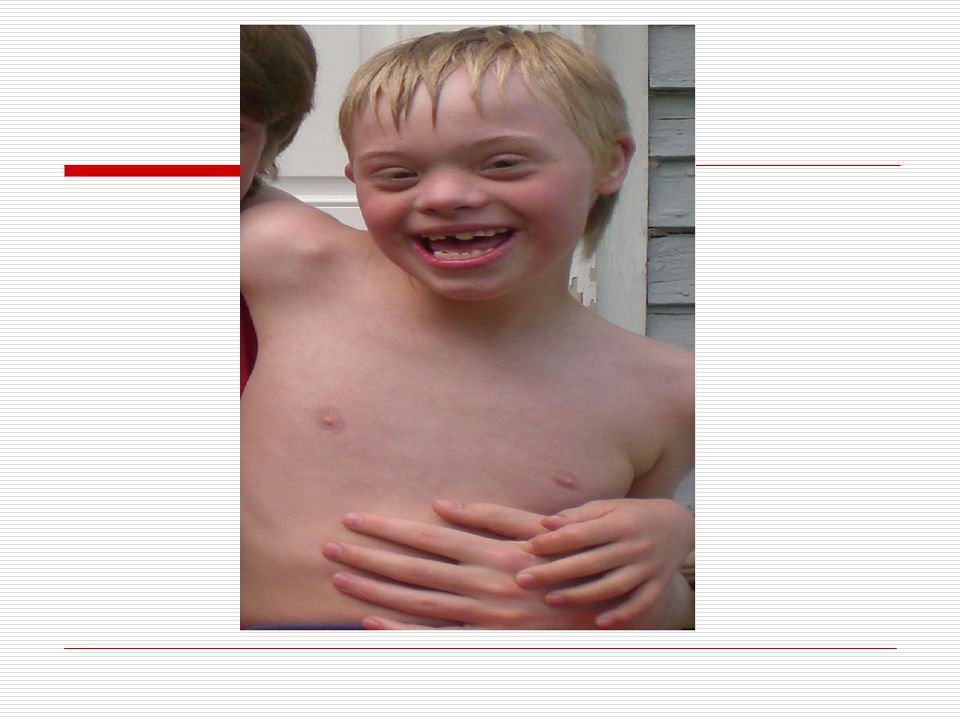
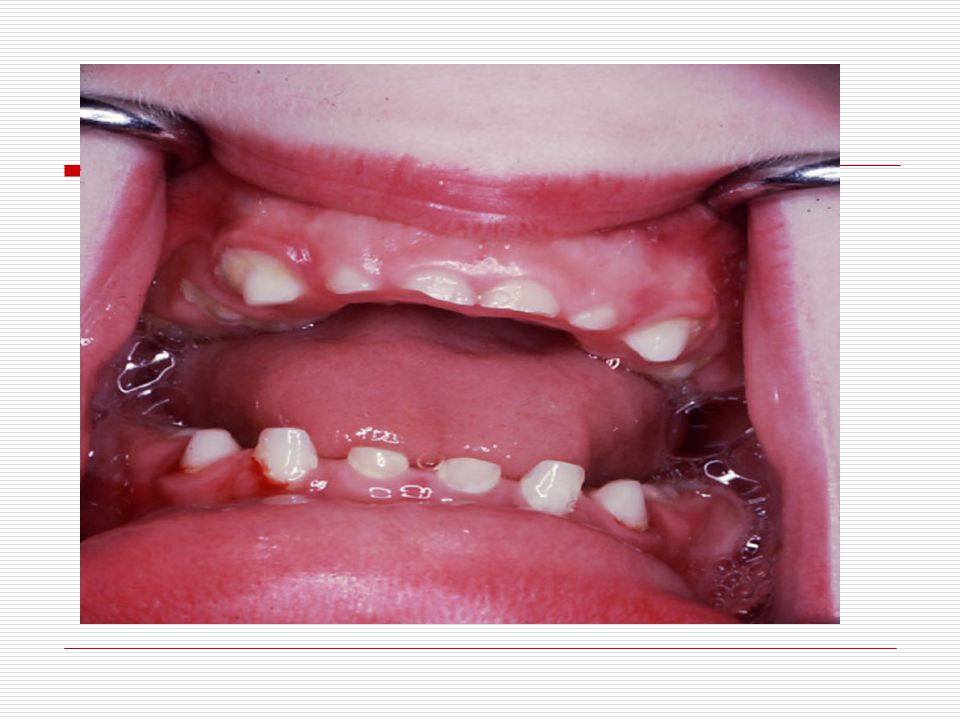
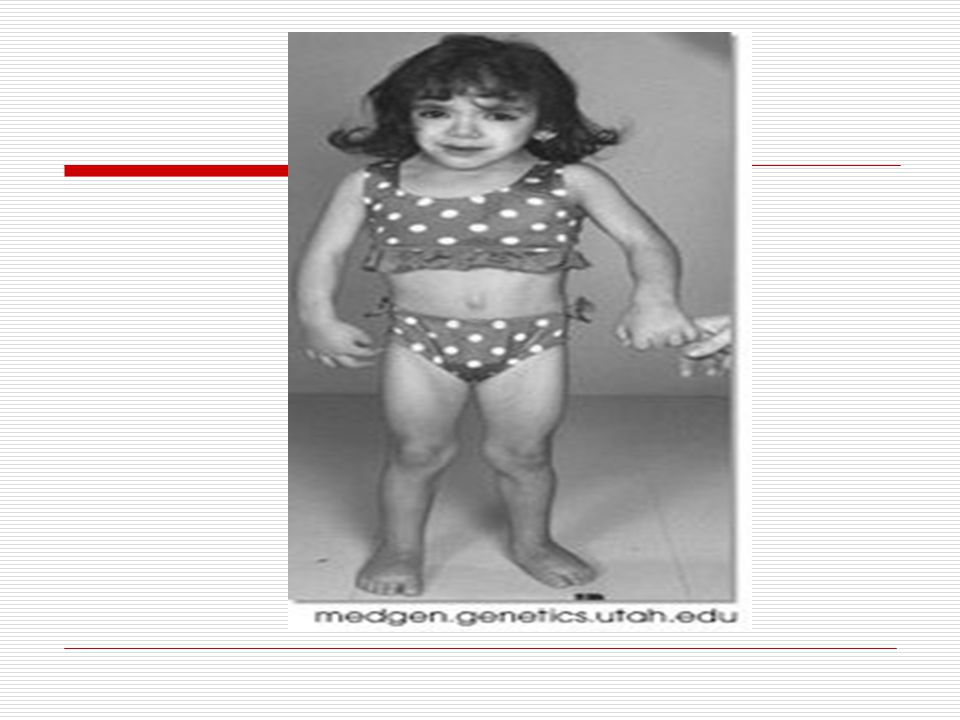
V . DOWN SENDROMU: İlk kez 1866 da Longdon DOWN tarafından mongoloid görünümde ve zeka geriliği ile birlikte bulunan bir sendrom olarak bulunmuştur. 21. kromozomun yer değiştirmesi veya birleşmemesinden dolayı meydana gelmektedir. 3 tip down sendromu vardır. TRİSOMY (regüler tip) MOZAİK TİP TRANSLOCATİON (translokasyon tip) Down sendromluların başlıca özellikleri;
 MOZAİK TİP. TRANSLOCATİON (translokasyon tip) Down sendromluların başlıca özellikleri;")
25
Baş; vücuda oranla nispeten ufaktır. Art kafa, yassı görünür
Baş; vücuda oranla nispeten ufaktır. Art kafa, yassı görünür. Kafatası yapısı “brakisefalik”tir (kafatası indisi’nin 81 den büyük olması ve doğal olarak kafatasının arkaya doğru basık oluşu). Alnın açık olması görülmektedir. Yüz; kendilerine özgü tipik yassı yüz görünümleri vardır. Ağız; genelde açıktır. Ağız kaslarının gevşekliği ve ağzın küçük olması dilin büyük olması nedeniyle bazılarında dil ağızlarından dışarı çıkar. Dişler bazı çocuklarda eksik ve düzensiz yerleşim görünür. Diş çıkartma gecikebilir. Dil; doğumda görünüşte normaldir. İleri yaşlarda ise dilde çanaksı kabarcıklar ve hipertrofi (dilde aşırı büyüme) görülür. Dillerinin üzeri kırışık olup, sıklıkla dil üzerinde merkez oluğu yoktur.
. Alnın açık olması görülmektedir. Yüz; kendilerine özgü tipik yassı yüz görünümleri vardır. Ağız; genelde açıktır. Ağız kaslarının gevşekliği ve ağzın küçük olması dilin büyük olması nedeniyle bazılarında dil ağızlarından dışarı çıkar. Dişler bazı çocuklarda eksik ve düzensiz yerleşim görünür. Diş çıkartma gecikebilir. Dil; doğumda görünüşte normaldir. İleri yaşlarda ise dilde çanaksı kabarcıklar ve hipertrofi (dilde aşırı büyüme) görülür. Dillerinin üzeri kırışık olup, sıklıkla dil üzerinde merkez oluğu yoktur.")
26
Gözler birbirinden ayrıktır ve çekik gözlüdürler
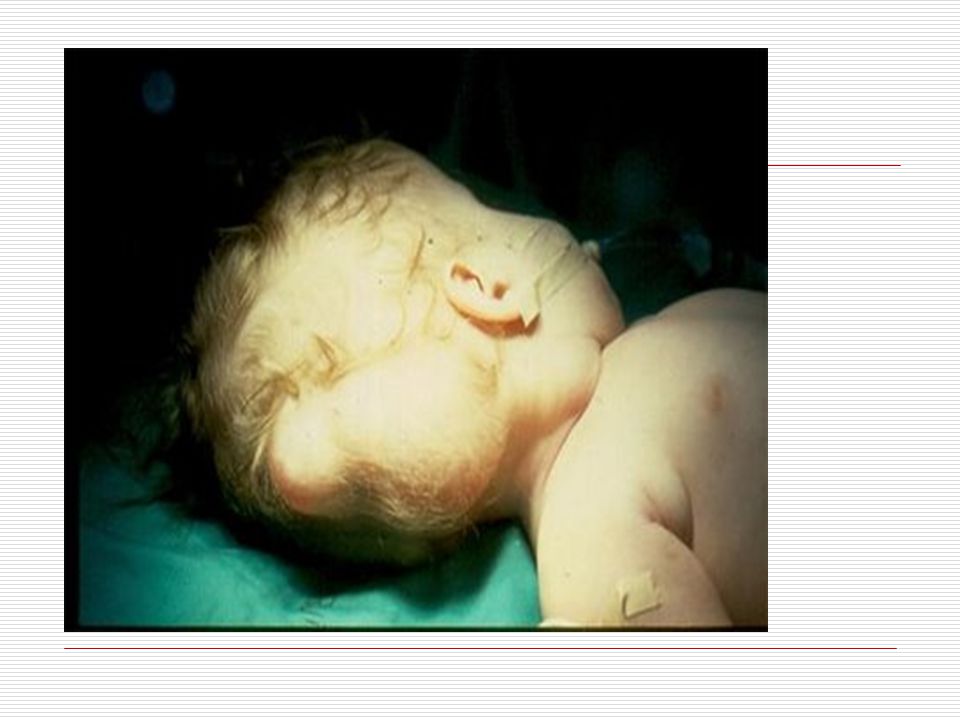
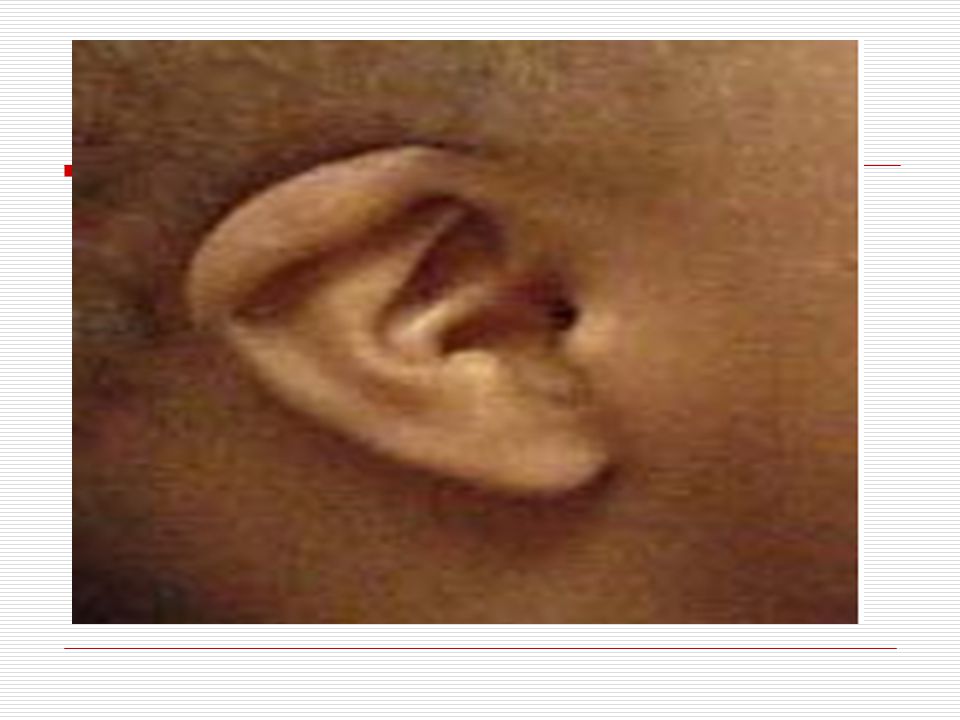
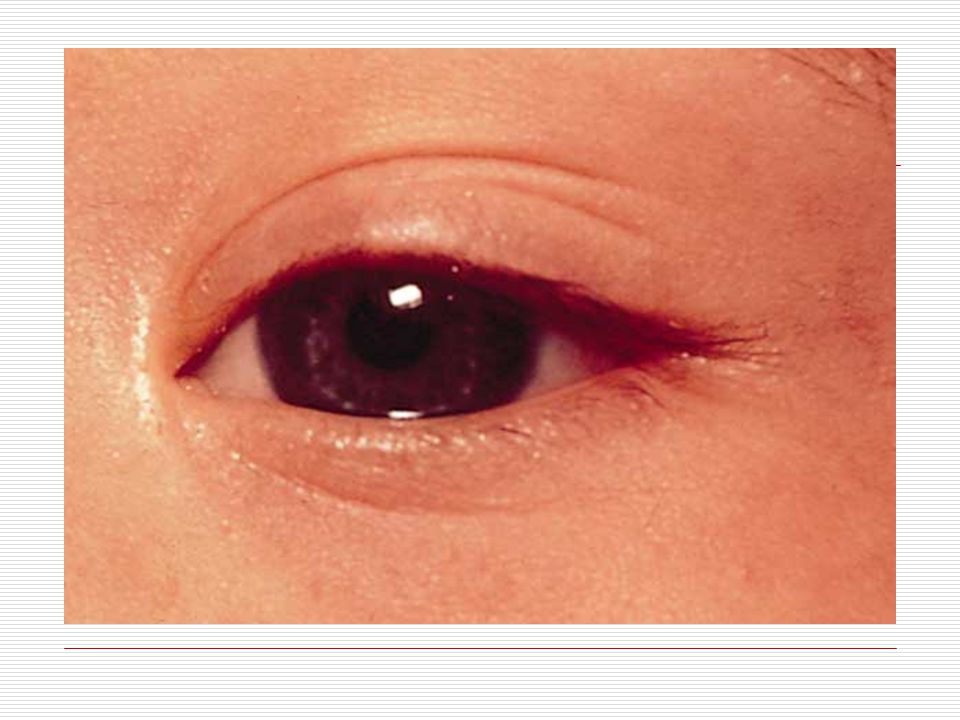
Gözler birbirinden ayrıktır ve çekik gözlüdürler. Yeni doğan döneminde, göz içinde tuz taneleri gibi görülen gri- beyaz lekeler görülebilir. Göz kapaklarında kıvrım mevcuttur. Çocuk büyüdükçe bu kıvrımlarda belirginleşir. Strabismus, nistagmus, katarakt gibi görme sorunları ortaya çıkabilir. Burun kökü düzleşmiş gibidir. Bunun için burunları basık gibidir. Genellikle burun iltihabı görülür. Kulaklar; kafada normalden daha küçük ve düşük bir seviyede durur. Bazılarının kulak kepçelerinin üst tarafı aşağı doğru sarkıktır. Antelis ( dış kulağın iç kenarı) az gelişmiştir. Kulak memeleri nispeten küçüktür.
 az gelişmiştir. Kulak memeleri nispeten küçüktür.")
27
Saçlar; Çocuklarda düz, seyrek ve yumuşaktır ileri yaştakilerde ise sert ve kabacadır. Sakallar zayıf ve belli belirsizdir. Erişkinlerde koltukaltı ve cinsel organ yakınındaki kılar kıt ve düzdür. Ense; kısa ve geniştir. Ense cildi gevşek olduğundan, ensede genellikle katlar vardır. Saç çizgisi normalden daha aşağıdadır. Göğüs: Bazı çocuklarda güvercin göğüs olabilir. Ergenlik esnasında memeler küçük kalır. Ama erişkinlikte normal olabilir. Bütün yaşlarda meme uçları küçüktür. Areola ( meme etrafındaki kahverengi kısım) belli belirsizdir.
 belli belirsizdir.")
28
Elleri küçük ve geniş, el parmakları kısa ve künttür
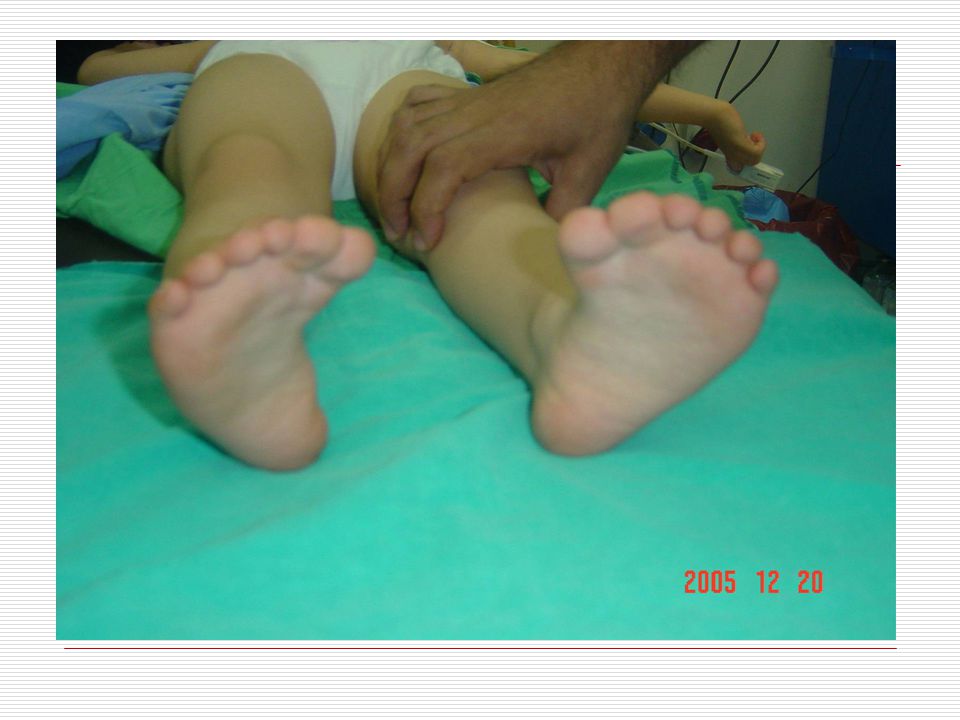
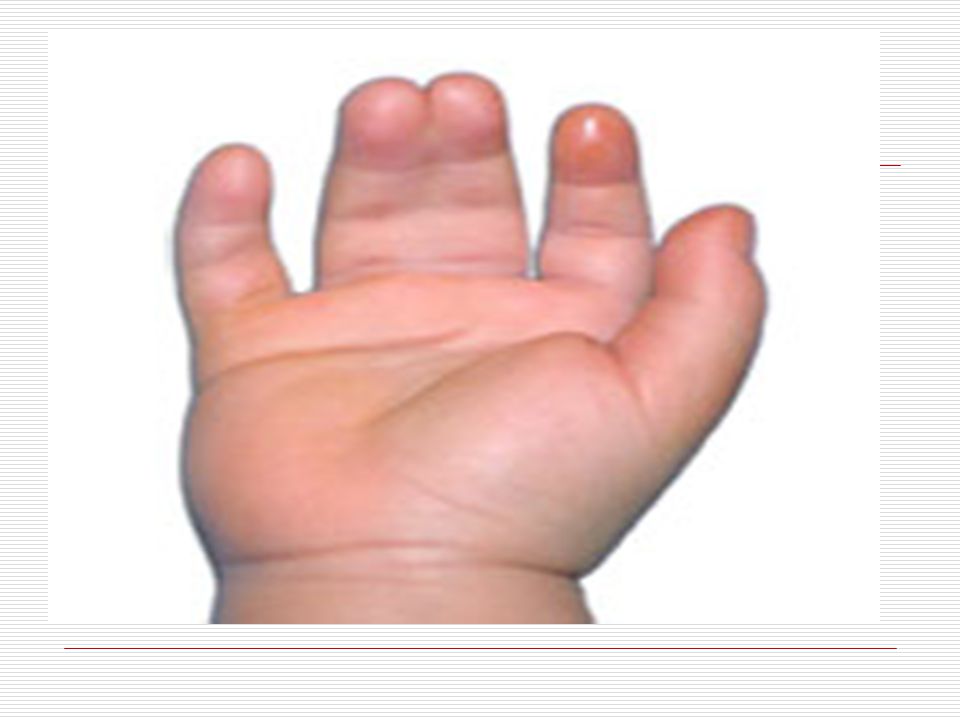
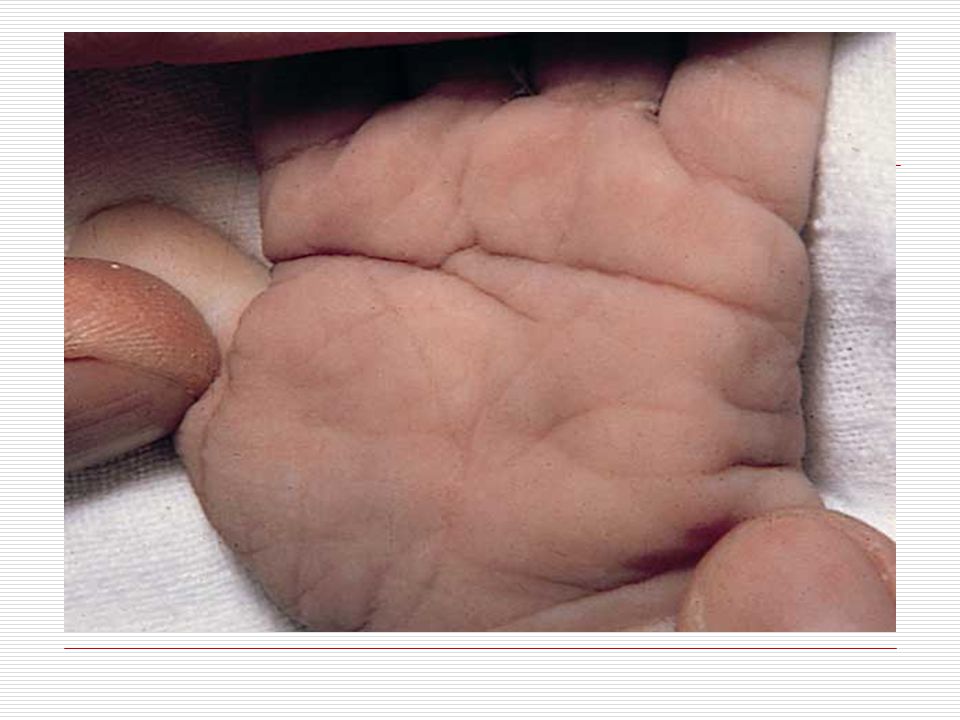
Elleri küçük ve geniş, el parmakları kısa ve künttür. Ellerin serçe parmakları genellikle içe doğru eğimlidir (klinodaktili), iç bükeyleşme vardır. Bunun nedeni bu parmağın orta boğumunun az gelişmiş olmasıdır. Bazılarında dört parmağın altında enlemesine tek bir tane kalın bir çizgi vardır. Ayak 1. ve 2. Parmak arası bazen aralıktır (Sandalet boşluğu). Ayak tabanında geriye topuğa doğru uzanan kırışıklık mevcuttur. El veya ayakta fazla parmak görülebilir (Polidaktili). Özellikle çocuklarda üst ve aşağı uzuvlarda eklemlerde genellikle hyperextensible (deride genişlik ve aşırı bolluk) görülür.
, iç bükeyleşme vardır. Bunun nedeni bu parmağın orta boğumunun az gelişmiş olmasıdır. Bazılarında dört parmağın altında enlemesine tek bir tane kalın bir çizgi vardır. Ayak 1. ve 2. Parmak arası bazen aralıktır (Sandalet boşluğu). Ayak tabanında geriye topuğa doğru uzanan kırışıklık mevcuttur. El veya ayakta fazla parmak görülebilir (Polidaktili). Özellikle çocuklarda üst ve aşağı uzuvlarda eklemlerde genellikle hyperextensible (deride genişlik ve aşırı bolluk) görülür.")
29
Deri: Eller ve ayaklar çoğunlukla beneklidir
Deri: Eller ve ayaklar çoğunlukla beneklidir. Deri, bebeklikte yumuşak ve gevşek, erişkinlikte ise kaba ve sert’tir. Bazen çocuklarda siyanoz (ciltte morarma) görülür. Bazı çocukların yanakları genellikle kırmızı ve pulludur. Boyları genellikle kısadır.
 görülür. Bazı çocukların yanakları genellikle kırmızı ve pulludur. Boyları genellikle kısadır.")
40
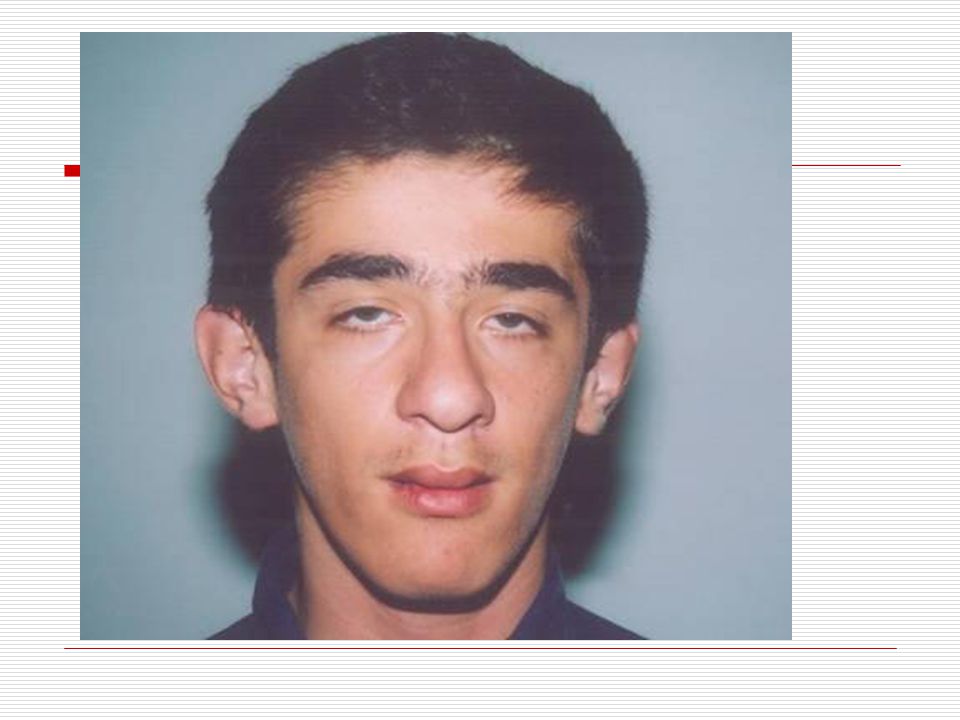
VI. FRAJİL X SENDROMU: Frajil X sendromu genetik bir hastalıktır. FMR1 adı verilen gen başlangıçta yinelenir (60’a kadar normal). Frajil X sendromlu kişilerde bu yinelenme kısmı normalden daha büyük bir şekilde veya daha fazla sayıda olur ( 200 den fazla yinelenmesi ). Bunun sonucunda ise Frajil X sendromu meydana gelir. Frajil X erkeklerde kızlara oranla daha çok görülür. Erkeklerin hafiften ağıra kadar değişebilen yetersizlik görülebilir. Kızlarda ise neredeyse çoğu normal zekaya sahiptir. Ancak üçte birinde hafif düzeyde yetersizlik görülür. Frajil X sendromlu çocuklarda bir takım fiziksel özellikler görülür. Bunların en önemlileri şunlardır:
. Frajil X sendromlu kişilerde bu yinelenme kısmı normalden daha büyük bir şekilde veya daha fazla sayıda olur ( 200 den fazla yinelenmesi ). Bunun sonucunda ise Frajil X sendromu meydana gelir. Frajil X erkeklerde kızlara oranla daha çok görülür. Erkeklerin hafiften ağıra kadar değişebilen yetersizlik görülebilir. Kızlarda ise neredeyse çoğu normal zekaya sahiptir. Ancak üçte birinde hafif düzeyde yetersizlik görülür. Frajil X sendromlu çocuklarda bir takım fiziksel özellikler görülür. Bunların en önemlileri şunlardır:")
41
Yüz; ince ve uzun bir görünümü vardır.
Dudaklar; normalden büyük ve dışarı doğru çıkık dudaklara sahiplerdir. Yüksek damak görülür. Kulaklar; büyük ve belirgin bir şeklidedir. Alın; açık geniş bir alna sahiplerdir. Çene yapıları aşağı doğru sivrileşen haldedir. Kafaları normalden daha büyüktür. İri testisler (ergenlik sonrası) Özellikler birincil olarak görülür.
 Özellikler birincil olarak görülür.")
42
Her çocukta olmayabilen ikincil özellikler ise;
Düz tabanlık, çift eklemli parmak, iskelet bozuklukları, eklemlerde kolay ve aşırı bükülme, kaslarda gevşeklik, skolyoz (omurgada eğrilik), içeri çökük göğüs kafesi özellikleri görülebilir.
, içeri çökük göğüs kafesi özellikleri görülebilir.")
47
VII. CORNELLA DE LANGE SENDROMU:
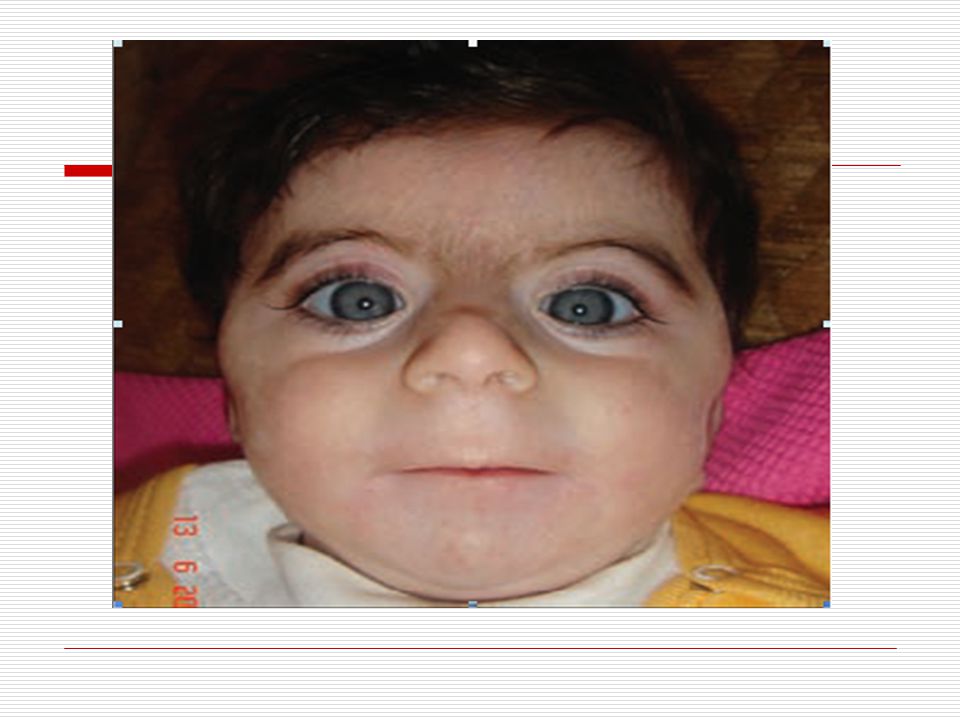
Brachman- de Lange sendromu olarak ta bilinen Cornelia de Lange sendromu’nun genetik sebepleri günümüzde hala araştırılmaktadır. Cornelia de Lange (CDL) sendromu nadir görülen, büyüme geriliği, mental retardasyon, tipik yüz görünümü, orta hatta birleşen kalın kaşlar (synophris), dar ince aşağı dönük üst dudak, mikromeli (kol ve bacakların aşırı kısa ve küçük oluşu) ile karakterize bir sendromdur. Bazı fiziksel özellikleri;
 sendromu nadir görülen, büyüme geriliği, mental retardasyon, tipik yüz görünümü, orta hatta birleşen kalın kaşlar (synophris), dar ince aşağı dönük üst dudak, mikromeli (kol ve bacakların aşırı kısa ve küçük oluşu) ile karakterize bir sendromdur. Bazı fiziksel özellikleri;")
48
Kafa; genellikle mikrosefali görülür.
Yüz’e ait en sık gözlenen karakteristik özellikler; ince aşağı doğru dönmüş dudaklara sahiptirler. Ağız; bazı çocuklarda yarık damak görülebilir. Kulakları normal seviyeden daha aşağıda yer alır Kirpikleri uzundur Kaşlar çoğunlukla ortada birleşir (sinofriz) ve kalın bir kaş yapısına sahiplerdir.
 ve kalın bir kaş yapısına sahiplerdir.")
49
Burun; bir çoğunda antevert burun delikleri görülür.
El parmaklarında mikromeli, bilateral simian çizgisi görülebilir. El ve ayak parmaklarında klinodaktili (parmakların içe doğru kıvrık olması) görülebilir. Vücutlarında aşırı kıllanma mevcuttur. Boyları çoğunlukla normalden kısa olmaktadır. Çoğunlukla düşük kiloda doğarlar (düşük doğum ağırlığı)
 görülebilir. Vücutlarında aşırı kıllanma mevcuttur. Boyları çoğunlukla normalden kısa olmaktadır. Çoğunlukla düşük kiloda doğarlar (düşük doğum ağırlığı)")
53
VIII. WİLLİAMS SENDROMU:
İlk kez 1961 yılında Williams Beuren tarafından tanımlanmıştır. Yaklaşık doğumda bir görülen bir hastalıktır. 7. kromozomun bir kısmın yokluğu ile oluşur. Anne babadan geçen kalıtımsal bir hastalık değildir. Hastalığın varlığı kan testi ile saptanabilir. Williams sendromlu çocukların bazı fiziksel görünüm özellikleri aşağıdaki gibidir; Küçük birbirinden uzak gözler Küçük çene Yukarı kalkık burun Büyük ağız ve kalın dudaklar Düzensiz ve aralıklı dişlere sahiptirler. Anne babaya göre genellikle daha kısa boylu olurlar.

59
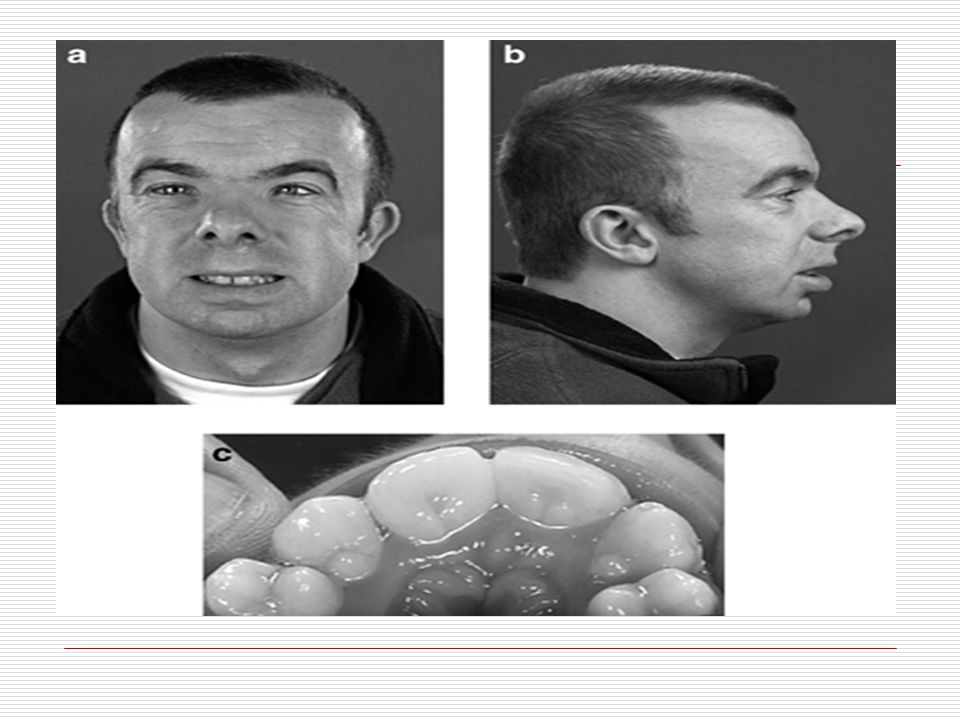
IX. RUBİNSTEİN TAYBİ SENDROMU:
Rubinstein–Taybi sendromu, geniş el ve ayak başparmakları, gaga burunla birlikte olan karakteristik yüz görünümü, kısa boy ile zihinsel gerilik ve motor becerilerdeki zayıflık kombinasyonundan oluşan nadir bir sendromdur. Oldukça nadir görülen bu sendromun tanısı radyolojik ve klinik bulgularla konur. Kafa: Bu hastaların çoğu mikrosefalidir. Ağız: Damak dar ve yukarıdan aşağı doğru bükülmüş gibidir. Dişlerin düzensiz yerleşmesi görülebilir.

60
Göz: Genel olarak gözlerde stabismus ve katarakt gibi sorunlar görülür.
Burun: Genel olarak gaga şeklinde bir burun yapıları vardır. Kulak bazılarında normalden düşük olabilir. Yüz: Bu hastalarda çoğunlukla hypertelorism (gözler arası açıklık) görülür. Gözler aşağı doğru eğiktir. Kaşlar kalındır ve genellikle ortada birleşir. Aynı zamanda regrognatizm (alna göre çenenin geride bulunması) görülür. Genellikle yüzlerini buruşturarak gülerler. Bazı hastalarda burun boşluğunu ikiye ayıran bölümün sapması veya sarkması görülebilir.
 görülür. Gözler aşağı doğru eğiktir. Kaşlar kalındır ve genellikle ortada birleşir. Aynı zamanda regrognatizm (alna göre çenenin geride bulunması) görülür. Genellikle yüzlerini buruşturarak gülerler. Bazı hastalarda burun boşluğunu ikiye ayıran bölümün sapması veya sarkması görülebilir.")
61
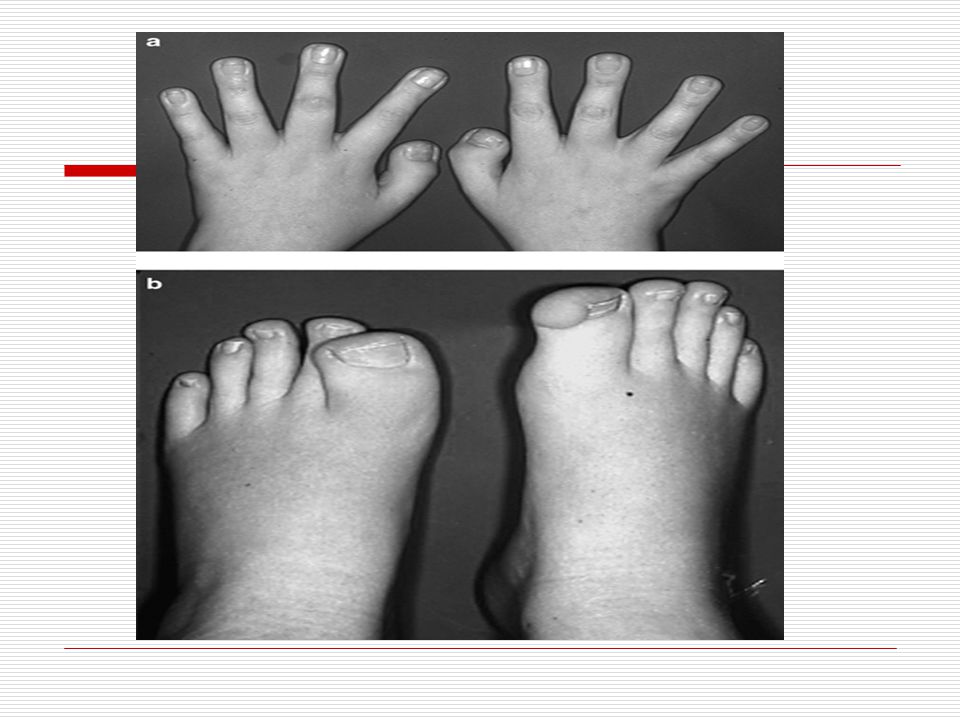
Cilt: Genel olarak alında, boynun arkasında kılcal damar tümörü görülebilir.
Saç: Bir çok hastanın vücudunun üstünde aşırı miktarda esmer kıllar vardır. El ve ayak başparmakları normalden daha geniştir. Ellerde sindaktili ve klinodaktili görülebilir. Ağırlık: Bazı hastaların normalden daha az doğum ağırlığı vardır.

66
X. TUBERUS SCLEROSİS (Tüberoskleroz) :
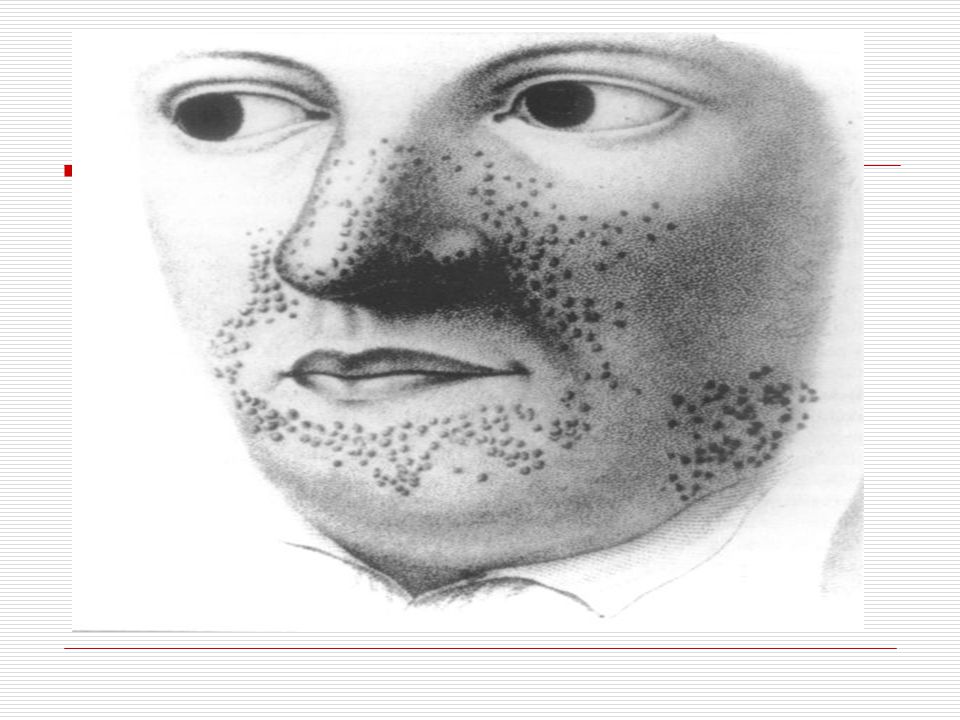
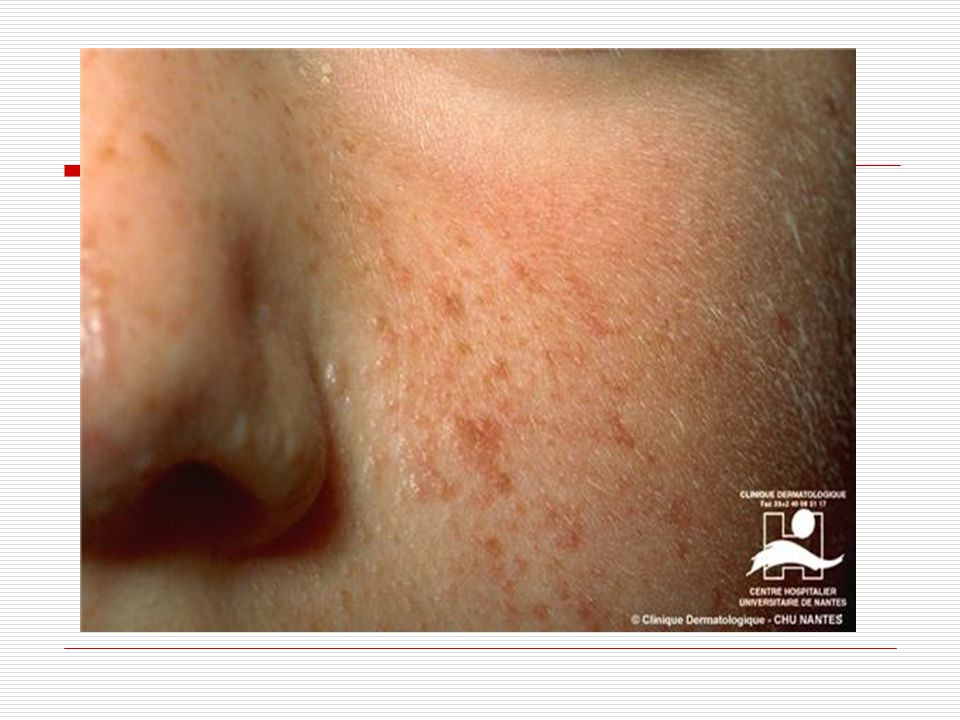
Beynin, gözlerin, derinin, kalbin, böbreklerin, akciğerlerin ve kemiklerin etkilenebildiği, otozomal dominant kalıtım gösteren nörokutanöz sendromdur . Klasik olarak nöbet, zeka geriliği ve çeşitli deri yaralarından oluşan bir üçleme ile tanımlanmaktadır. Etkilenen kişilerin üçte ikisinden fazlasında zihinsel gerilik vardır. Bazı fiziki özellikleri aşağıdaki gibidir; Cilt: İlk olarak Hypomelanic macules görülür: Vücudun herhangi bir bölgesindeki deri üzerinde beyaz yada daha açık renkteki lekelerdir. Bu lekelerin nedeni derideki pigment yada melanin(cilde renk veren madde) eksikliğidir. 1 cm den daha büyük ve birkaç ayrı düzüne de vücudun herhangi bir bölgesinde görülebilmektedir. Şekil olarak oval veya çizgisel olabilir.
 eksikliğidir. 1 cm den daha büyük ve birkaç ayrı düzüne de vücudun herhangi bir bölgesinde görülebilmektedir. Şekil olarak oval veya çizgisel olabilir.")
67
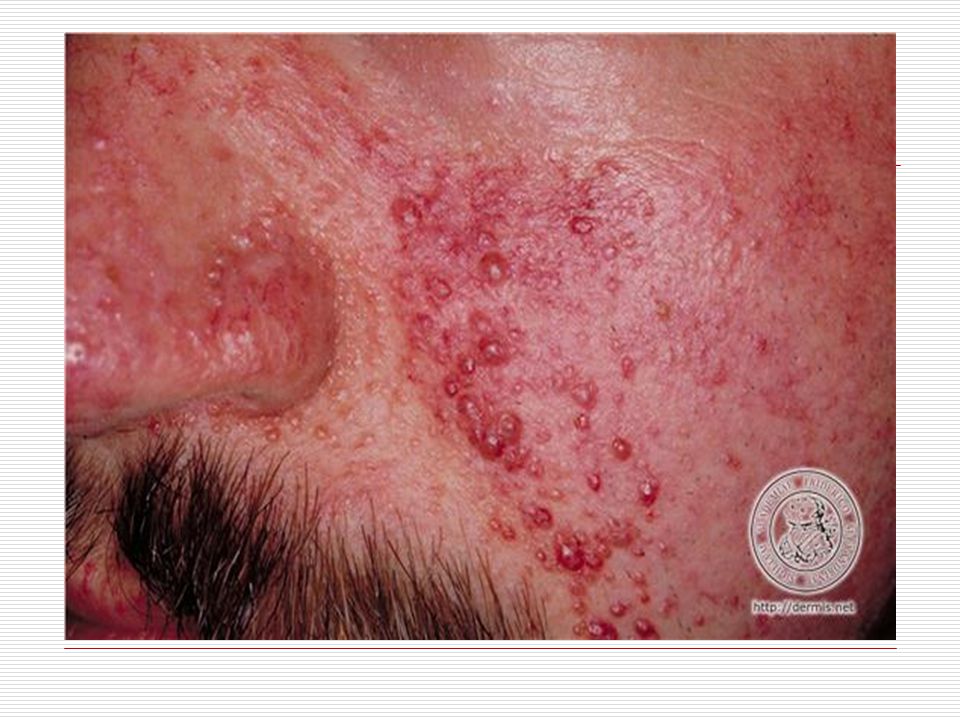
Hastaların %83’ünde adenoma sebaceum vardır
Hastaların %83’ünde adenoma sebaceum vardır. Bunlar 1 ile 7 mm arasında değişen et renkli veya pembe yumrulardır. Çoğunlukla yüzde ortaya çıkar ve akneye benzer. Genellikle 2 yaşından sonra ortaya çıkmakta zaman içinde daha belirgin olmaktadır. Yaşam boyu kalıcıdır. Shagreen patch görülür. Buda genellikle boyun ve sırt taki kalın, sert ve pütürlü alandır. Ayrıca bu çocuklarda yaprak tülü lekeleri görülür.

68
Gözler: Hastaların yarısında retinada lekeler vardır
Gözler: Hastaların yarısında retinada lekeler vardır. Bu lekeler çocuklukta mevcut olabilir. Bunlar küçük, yuvarlak ve biraz kabarıktır. Ağ tabakasının herhangi bir parçasında saptanabilir. El ve ayaklarda ungual fibroma (küçük etli tümör) görülür. Ayak tırnaklarının yada el tırnaklarının altında görülen tümörlerdir.
 görülür. Ayak tırnaklarının yada el tırnaklarının altında görülen tümörlerdir.")
75
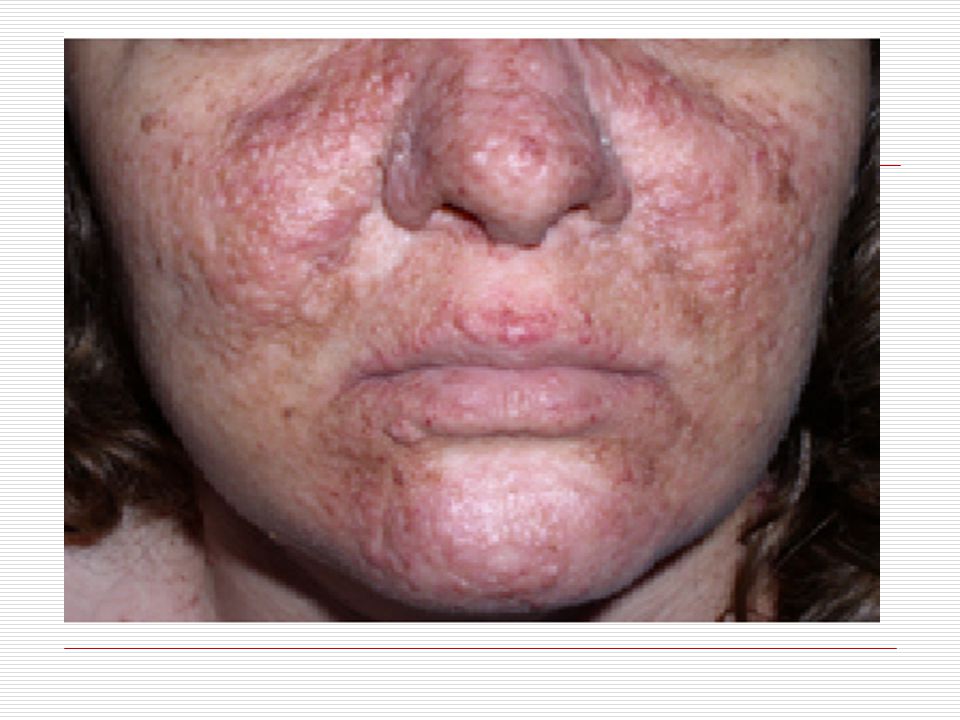
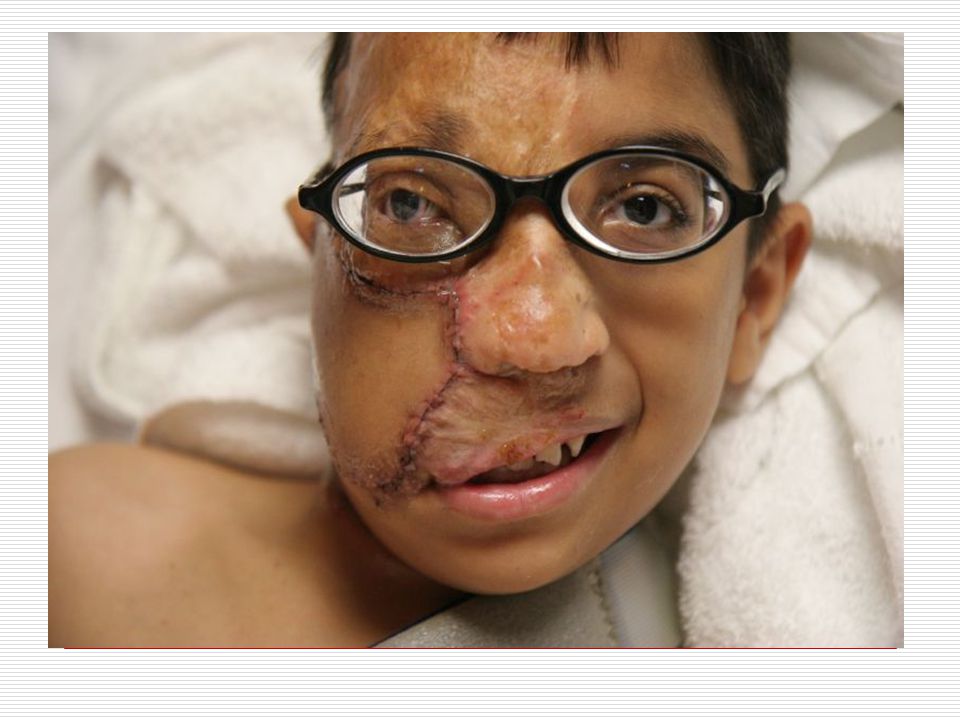
XI. STURGE-WEBER SENDROMU:
Hastalık yeni doğan veya erken çocukluk döneminde başlayan nöbetlerle ortaya çıkar. Bu hastalık kalıtsal değildir. Bu hastalık yüzde tek taraflı pembe-mor veya şarap kırmızısı yaralar (port-wine stain) ile özdeşleşir.Ağır vakalarda mental retardasyon ve kontralateral hemiparazi (beyinin etkilenen bölgesinin çaprazındaki vücut yarısındaki kuvvet kaybı) gözlenir. Bu hastaların bazı özellikleri;
 ile özdeşleşir.Ağır vakalarda mental retardasyon ve kontralateral hemiparazi (beyinin etkilenen bölgesinin çaprazındaki vücut yarısındaki kuvvet kaybı) gözlenir. Bu hastaların bazı özellikleri;")
76
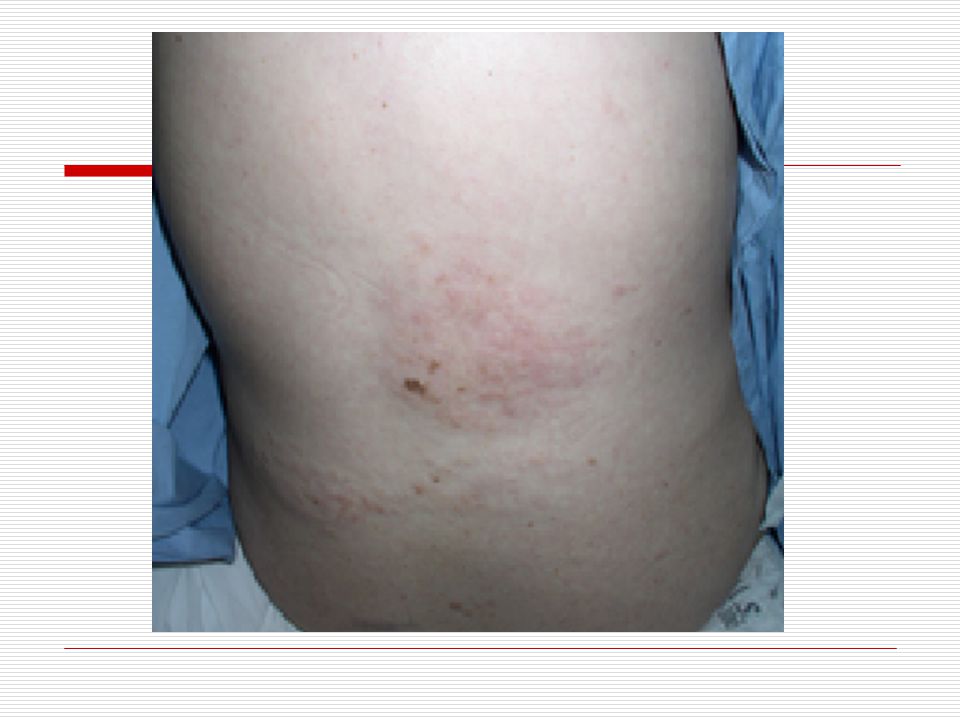
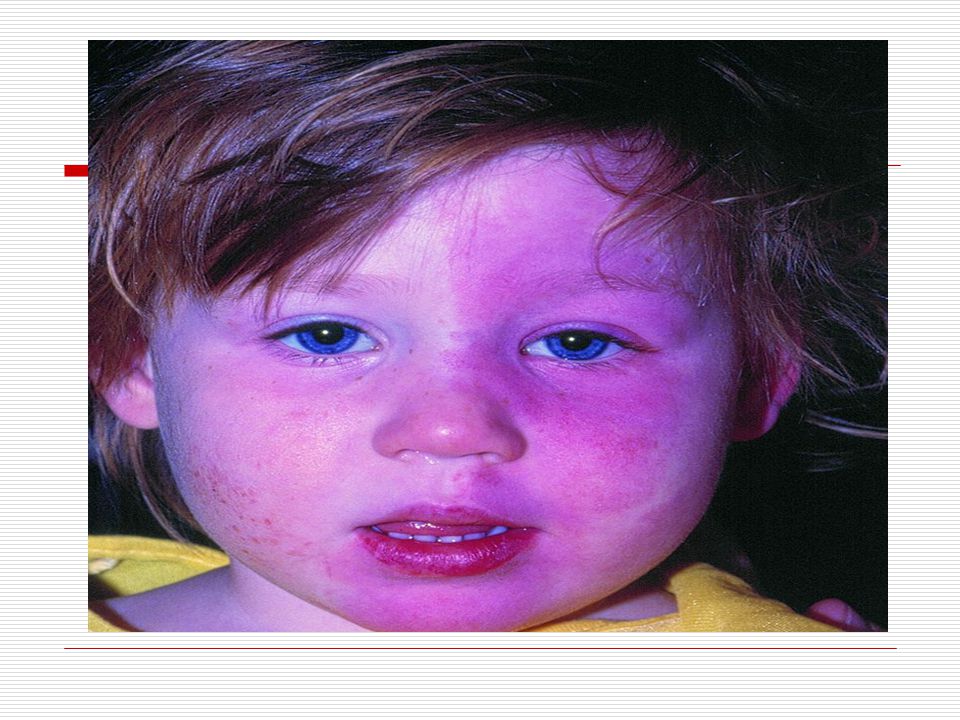
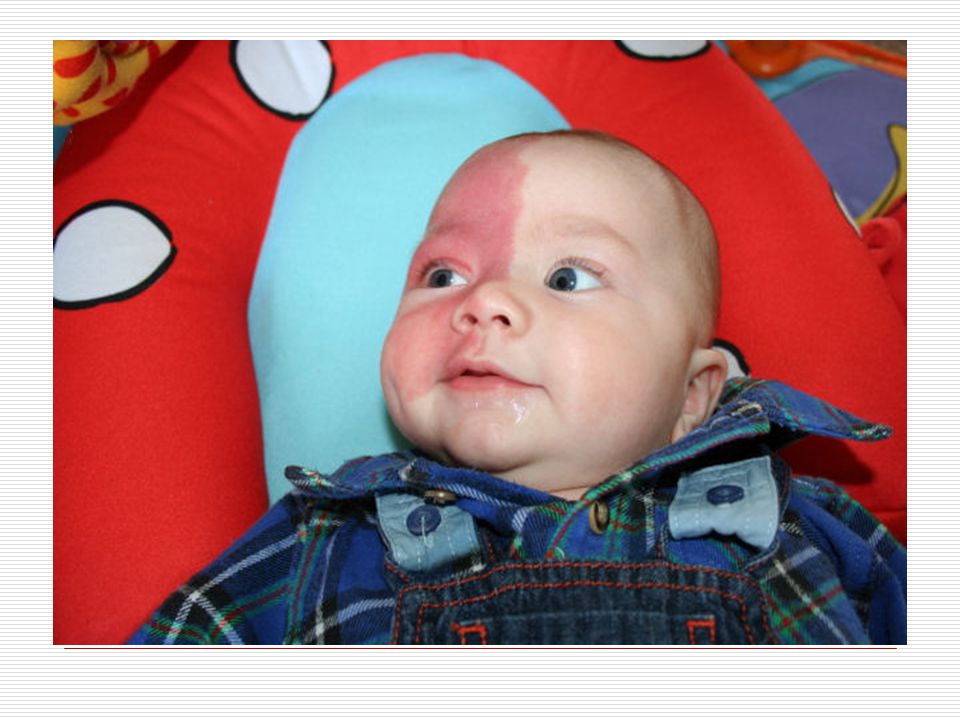
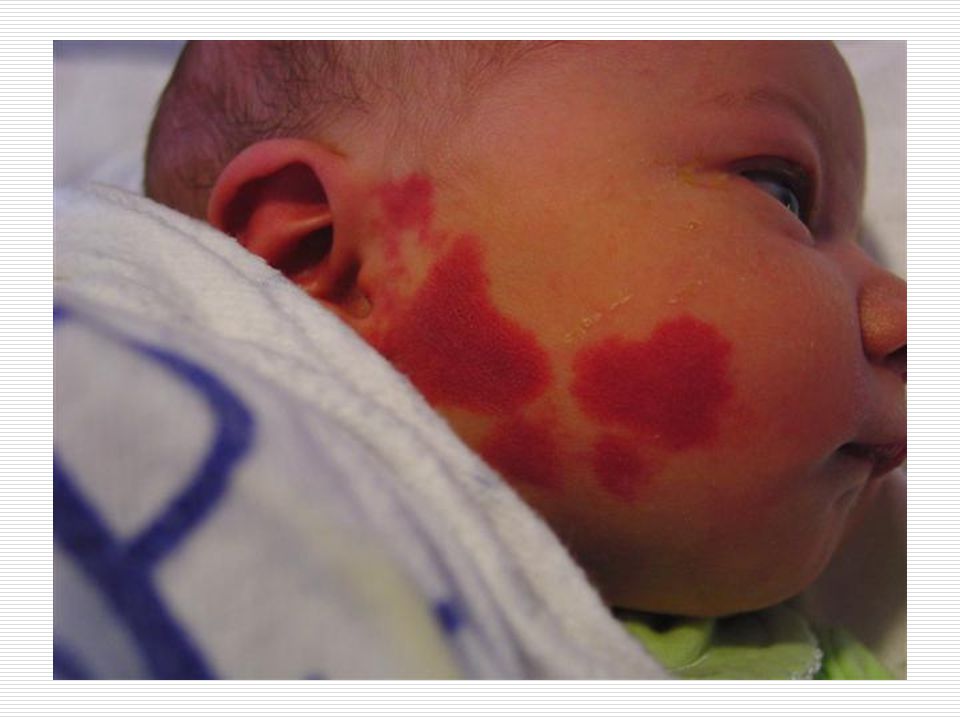
Cilt: Yüz de doğumdan itibaren nevus flammeus lekesi görülür
Cilt: Yüz de doğumdan itibaren nevus flammeus lekesi görülür. Bu leke yinede vücudun diğer bölgelerinde üç yönlü görülebilir. Bu leke genellikle keskince ve düz bir şekilde ayrılır. Sadece bir kısmı kaplar. Genellikle doğumda ve yüzün üst bölgesinde tek taraflı olmaya eğilim gösterir. Yaş ilerledikçe lekenin kapladığı alan koyulaşır ve siğile benzeyen şeyler çıkabilir. Bu leke genellikle alın ve üst göz kapaklarına kadar ulaşır.

77
Göz; bir çok hastada glokom ( göz içi basıncının giderek yükselmesi nedeniyle görme sinirinin giderek zayıflaması sonucu ortaya çıkan görme bozukluğu) ortaya çıkar. Ağız; nevus flammeus lekesi çoğunlukla ağız, burun ve boğazın yakınlarında olur. Bu lekeli alanların büyüklüğü değişebilir. Bazı çocuklarda dişlerin erken çıkması görülebilmektedir.

83
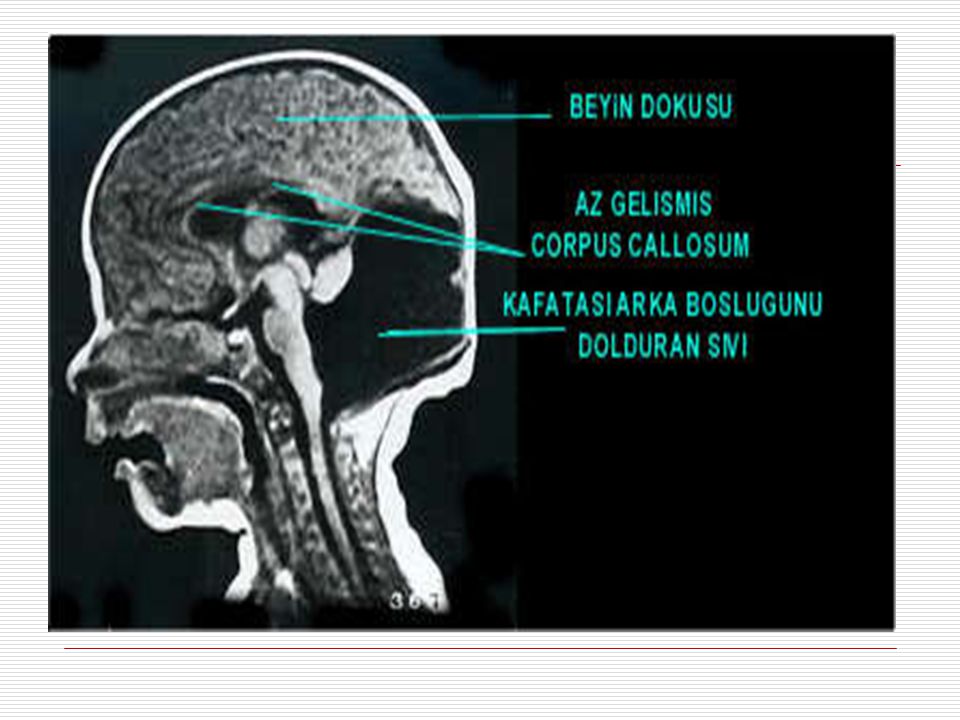
XII. DANDY-WALKER SENDROMU:
Hastalık dördüncü ventrikül denilen ve beyin omurilik sıvısının dolaştığı boşluklardan birinin doğuştan anormal genişlemesi, beyincikte iki beyincik yarımküresinin arasında yer alan ve vermis denilen bölümün yokluğu veya gelişiminin geri kalması ve bu anormallikler sonucunda kafatasının arka boşluğunda bir kist oluşması ile karakterizedir. Ayrıca hidrosefali yani kafa içi basıncının artması ve kafatasının genişlemesi de eşlik edebilir.

84
Baş; bazısında yeni doğduğunda başın büyüklüğü dikkat çekebilir
Baş; bazısında yeni doğduğunda başın büyüklüğü dikkat çekebilir. Bazısında ise doğduğunda başı normal olup yaşamın ilk yıllarında ani büyüme görülebilir. En bilindik özellikleri art kafanın çıkıntılı olmasıdır. Yüz; bu çocuklarda genellikle yüzde şekil bozuklukları görülür. Yarık dudak ve damak görülebilir. El ve ayaklarda polidaktili, sindaktili ve şekil bozuklukları görülebilir. Hemen hemen bütün dandy – walker sendromlu çocuklarda nöbetler görülür.

89
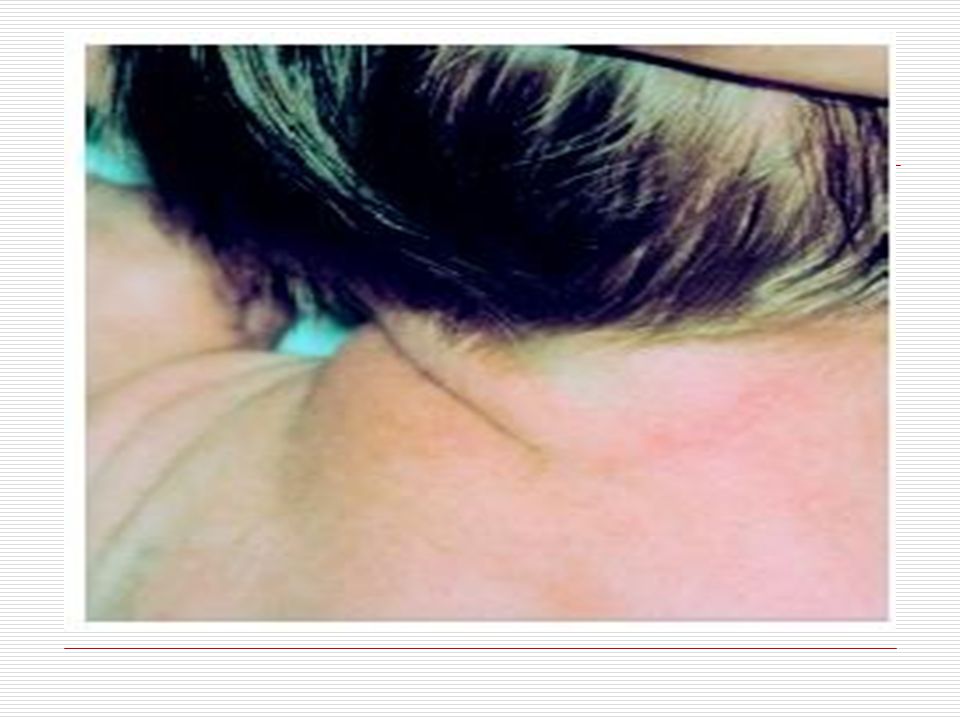
XIII. TURNER SENDROMU: Sadece kızlarda görülen ve bir X kromozomunun tamamen yada kısmen eksik olmasından kaynaklanır. Bazı özellikleri: Hastaların boylarının kısa olması Geniş bir göğsün olması ve memelerinin aralarında büyük boşluk olması Ense bölgesinde düşük saç çizgisi Düşük kulaklar Küçük alt çene

90
Perdeli boyun (boynun yan kısmıyla omuz üzeri arasındaki gevşek deri)
Sarkık göz kapakları Kaşık şeklinde tırnaklar Şiş eller ve ayaklar Kısa el ve ayak parmakları Kısırlık

Benzer bir sunumlar


























>")















