Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Temel Sitogenetik ve Nomenklatur; Dünden Bugüne Kromozom Analizleri
Prof. Dr. Seher Başaran İstanbul Üniversitesi, İstanbul Tıp Fakültesi, Tıbbi Genetik AD

2
Sitogenetiğin Tarihçesi
Kromozom,yunanca “renkli cisim” (“chroma + soma”) (1888, Waldeyer) Kalıtımın fiziksel temeli kromozomlardır (1892,Weissmann) Kalıtımın kromozomal teorisi ve kromozomların yapısını, fonksiyonlarını ve hastalık ilişkisini araştıran bilim dalı “sitogenetik” (1902, Sutton) Erkekler 47, dişiler 48 kromozom (1912, von Winiwarter) Kromozom sayısı 48 (1923, Painter) Kromozomlar boyandı (1924, Feulgen) Kolşisin (1932, Blakeslee ve Eigsti) Hipotonik solusyon kullanıldı (1953), Kolşisin+hipotonik şok kullanıldı (1955, Ford ve Hamerton) İnsanda 2n=46 (1956, Tijo ve Levan) “Modern sitogenetiğin başlaması” Down sendromu (1959, Lejeune) Ph kromozomu (1960, Nowell) “cat cry, 5p-” (1963, Lejeune) QFA (1968, Caspersson ) G band (1970, Drets veShaw ) ISCN (International System for Human Cytogenetic Nomenclature,1971) HRBT (Prometafaz, High Resolution Banding Technique, 1981) ISH (1981) FISH (1990, Lichter)
 (1888, Waldeyer) Kalıtımın fiziksel temeli kromozomlardır (1892,Weissmann) Kalıtımın kromozomal teorisi ve kromozomların yapısını, fonksiyonlarını ve hastalık ilişkisini araştıran bilim dalı sitogenetik (1902, Sutton) Erkekler 47, dişiler 48 kromozom (1912, von Winiwarter) Kromozom sayısı 48 (1923, Painter) Kromozomlar boyandı (1924, Feulgen) Kolşisin (1932, Blakeslee ve Eigsti) Hipotonik solusyon kullanıldı (1953), Kolşisin+hipotonik şok kullanıldı (1955, Ford ve Hamerton) İnsanda 2n=46 (1956, Tijo ve Levan) Modern sitogenetiğin başlaması Down sendromu (1959, Lejeune) Ph kromozomu (1960, Nowell) cat cry, 5p- (1963, Lejeune) QFA (1968, Caspersson ) G band (1970, Drets veShaw ) ISCN (International System for Human Cytogenetic Nomenclature,1971) HRBT (Prometafaz, High Resolution Banding Technique, 1981) ISH (1981) FISH (1990, Lichter)")
3
G Bantlama (GTG) GTG- Karyotip Metafaz plağı Prometafaz plağı
HRBT karyotip

4
~ 1 bant 6 milyon nükleotid (~6 Mb ve 10’larca gen)
Metafaz Prometafaz(HRBT) ~ 500 bant çözünürlükte ~ 1 bant 6 milyon nükleotid (~6 Mb ve 10’larca gen) bant/haploid set
 ~ 500 bant çözünürlükte. ~ 1 bant 6 milyon nükleotid (~6 Mb ve 10’larca gen) bant/haploid set.")
5
Fluoresan In Situ Hibridizasyon (FISH)
Fluorescence in situ hybridization (FISH) Need to suspect a specific diagnosis! Spektral karyotipleme In the early 1990s, recurrent small microdeletions of the genome not visible by light microscopy were identified associated with characteristic syndromes. These were only detectable by fluorescence in situ hybridization (FISH) or similar techniques and include the common microdeletion syndromes of: Wolf-Hirshhorn, Cri du Chat, Williams, Prader-Willi, Angelman, Rubinstein-Taybi, Miller-Dieker lissencephaly, Smith-Magenis, Alagille, DiGeorge and 22q11 deletion syndrome. Although each syndrome has characteristic distinguishing features all are also associated with varying degrees of mental retardation and in some cases associated with specific areas of intellectual impairment. Kromozom 21 Kromozom 13 Kromozom 18 Kromozom X Kromozom Y
 Need to suspect a specific diagnosis! Spektral karyotipleme. In the early 1990s, recurrent small microdeletions of the genome not visible by light microscopy were identified associated with characteristic syndromes. These were only detectable by fluorescence in situ hybridization (FISH) or similar techniques and include the common microdeletion syndromes of: Wolf-Hirshhorn, Cri du Chat, Williams, Prader-Willi, Angelman, Rubinstein-Taybi, Miller-Dieker lissencephaly, Smith-Magenis, Alagille, DiGeorge and 22q11 deletion syndrome. Although each syndrome has characteristic distinguishing features all are also associated with varying degrees of mental retardation and in some cases associated with specific areas of intellectual impairment. Kromozom 21. Kromozom 13. Kromozom 18. Kromozom X. Kromozom Y.")
6
~500 bant düzeyinde “Karyotipleme”
Tek test tüm genom/kromozomlar Tüm sayısal anomaliler; 47,XX,+ 21 69,XXX 45,X 47,XY,+13 .nuc ish(DXZ1x1) .nuc ish(D21S65x3) .nuc ish(RB1x3)
 .nuc ish(D21S65x3) .nuc ish(RB1x3)")
7
~500 bant düzeyinde “Karyotipleme” Yapısal kromozom anomalilerinin araştırılmasında sitogenetik yaklaşım 46,XX,5p- Bant paternine göre Karyotip;46,XX,5p- Sadece delesyon ? Derivatif kromozom ? Parental kromozom analizleri; Anne ve Baba “Normal” 46,XX,del(5)(p15.1->pter) dn FISH ile kırık noktalarının belirlenmesi ve konfirmasyon; 46,XX,del(5)(p15.1->pter).ishdel(5)(p15.1->pter)(CTNND2-) Yorum; Olgudaki 5p15.1->pter bölgesi için parsiyel monozominin fenotipi etkilemesi beklenmektedir.
(p15.1->pter) dn. FISH ile kırık noktalarının belirlenmesi ve konfirmasyon; 46,XX,del(5)(p15.1->pter).ishdel(5)(p15.1->pter)(CTNND2-) Yorum; Olgudaki 5p15.1->pter bölgesi için parsiyel monozominin fenotipi etkilemesi beklenmektedir.")
8
~500 bant düzeyinde “Karyotipleme”
Fetus 46,XY,3p+ ,der(3)t(3;20)(p25.3;p11.2) mat ,der(3)(20pter->20p11.2::3p25.3->3qter)mat Anne 46,XX,t(3;20)(p25.3;p11.2) Yorum; Fetus, annedeki translokasyonun Adjacent-1 dağılımı ürünü derivatif 3 kromozomunu taşıdığından 20pter->p11.2 bölgesi için trizomik ve 3p25.3->pter bölgesi için monozomiktir ve fenotipik olarak etkilenmesi beklenmektedir.
t(3;20)(p25.3;p11.2) mat. ,der(3)(20pter->20p11.2::3p25.3->3qter)mat. Anne 46,XX,t(3;20)(p25.3;p11.2) Yorum; Fetus, annedeki translokasyonun Adjacent-1 dağılımı ürünü derivatif 3 kromozomunu taşıdığından 20pter->p11.2 bölgesi için trizomik ve 3p25.3->pter bölgesi için monozomiktir ve fenotipik olarak etkilenmesi beklenmektedir.")
9
Kompleks Kromozom Anomalilerinin Aydınlatılması 46,XY,t(2q;8q;13q)?
46,XY,t(8;13)(q24.13;q21.2).ish ins(2)(p16.2q33.2q22.2)(wcp2, wcp8, wcp13, 2pqsubtel).ish ins(2)(pter->p16.2::q33.2->q22.2::p16.2->q33.2->qter)dn Karaman et al. 2009 Yorum;Mikroskobik olarak dengeli görünen bu de novo kompleks kromozom anomalisinin fenotip üzerine etkisinin a-CGH tekniği ile araştırılması gerekmektedir. 2pq subtel 2pq wcp
(q24.13;q21.2).ish ins(2)(p16.2q33.2q22.2)(wcp2, wcp8, wcp13, 2pqsubtel).ish ins(2)(pter->p16.2::q33.2->q22.2::p16.2->q33.2->qter)dn Karaman et al Yorum;Mikroskobik olarak dengeli görünen bu de novo kompleks kromozom anomalisinin fenotip üzerine etkisinin a-CGH tekniği ile araştırılması gerekmektedir. 2pq subtel. 2pq wcp.")
10
~500 bant düzeyinde “Karyotipleme”
Klinik yönlendirme varsa bilinen mikrodelesyon sendromları (~ 3-5Mb) Klinik yönlendirme varsa bilinen mikrodelesyon sendromları (~ 3-5Mb) A B A. 46,--,del(22)(q11.2) . ishdel(22)(q11.2q11.2)(D22S75-) Yorum; Olguda saptanan 22q11.2 bandındaki delesyonun fenotipi etkilemesi beklenmektedir. Bu delesyon DGS/VCFS ile uyumludur. B. 46,--(D22S75x2) Yorum; FISH incelemesinde D22S75 probu ile delesyon saptanmamıştır.
 Klinik yönlendirme varsa bilinen mikrodelesyon sendromları (~ 3-5Mb) A. B. A. 46,--,del(22)(q11.2) . ishdel(22)(q11.2q11.2)(D22S75-) Yorum; Olguda saptanan 22q11.2 bandındaki delesyonun fenotipi etkilemesi beklenmektedir. Bu delesyon DGS/VCFS ile uyumludur. B. 46,--(D22S75x2) Yorum; FISH incelemesinde D22S75 probu ile delesyon saptanmamıştır.")
11
Marker kromozom: Çoğunlukla normal kromozom setine ilave, rutin bantlama teknikleri ile tanınamayan kromozomlar 47,XY,+mar 47,XY,+idic(15)(q12)(D15Z4++,SNRPN++) Yorum; 15. kromozom kökenli, izodisentrik ilave kromozom, olguda 15p->15q12 için tetrazomiye yol açmaktadır ve fenotipi etkilemesi beklenmektedir. Marker kromozomların yarısı fenotipi etkilemeyen ökromatin taşımayan kromozomlar olduğundan kromozomal kökeninin ve içeriğinin belirlenmesi gerekir (FISH veya a-CGH) !!!! D15Z4 47,XY,+mar SNRPN
(q12)(D15Z4++,SNRPN++) Yorum; 15. kromozom kökenli, izodisentrik ilave kromozom, olguda 15p->15q12 için tetrazomiye yol açmaktadır ve fenotipi etkilemesi beklenmektedir. Marker kromozomların yarısı fenotipi etkilemeyen ökromatin taşımayan kromozomlar olduğundan kromozomal kökeninin ve içeriğinin belirlenmesi gerekir (FISH veya a-CGH) !!!! D15Z4. 47,XY,+mar. SNRPN.")
12
Kromozomal Polimorfizmler
1, 9, 16 ve Y ve tüm akrosentrik kromozomlarda sentromer, satellit bölgelerinde görülen uzama, duplikasyon, inversiyon, kısalma gibi özellikler “polimorfik bulgu” olarak tanımlanır ve toplumda yaygındır, sorun yaratmaz. Örn; inv9, Yqh++, 13ps++ Ender görülen heterokromatin bölge değişiklikleri ise yanlış tanıya götürebilir. Bu nedenle değişimin fenotip üzerine etkisi diğer tekniklerle araştırılmalı ve doğru tanıya gidilmelidir.

13
Polimorfizm ya da yapısal anomali??
46,--,22p+. ishdup(22)(q11.1->q13.1) (wcp22, N25x2) Yorum; duplikasyon 22q11.1->q13.1, Fenotipi etkilenmesi beklenmektedir. 46,--,22ps++ Yorum; polimorfik bir özellik ve fenotipi etkilemesi beklenmemektedir
(q11.1->q13.1) (wcp22, N25x2) Yorum; duplikasyon 22q11.1->q13.1, Fenotipi etkilenmesi beklenmektedir. 46,--,22ps++ Yorum; polimorfik bir özellik ve fenotipi etkilemesi beklenmemektedir.")
14
Mozaisizm Tanım: Bir organizmada aynı bir zigottan kaynaklanan farklı genetik yapıda hücrelerin bulunmasıdır. Mozaisizm, kromozom anomalileri ya da tek gen mutasyonları için olabilir. Bir doku ile de sınırlı kalabilir. Fetal Kromozom Analizlerinde Karşılaşılan Mozaisizmler ve Sınıflandırması: 1. Psödomozaisizm (yalancı): Fetusta bulunmayan a. Tek kapta/klonda “Tek hücre mozaisizmi (SCM)”- 1. düzey b. Tek kapta “Çok hücre mozaisizmi (MCM)”- 2. düzey 2. >2 kap/klonda “Gerçek mozaisizm” - 3. düzey Aynı bir kromozom anomalisinin; farklı kültür kaplarında (“flask tekniği) ya da farklı preparatlarda (in situ tekniği) >2 metafazda (monozomilerde >3 metafazda) saptanması ile “gerçek mozaisizm” tanısı konur. mos 45,X [25]/47,XXX [25]/46,XX [15] Mozaik Turner sendromu mos 46,XY [25]/47,XY,+21 [25 ] Mozaik Down sendromu
: Fetusta bulunmayan. a. Tek kapta/klonda Tek hücre mozaisizmi (SCM) - 1. düzey. b. Tek kapta Çok hücre mozaisizmi (MCM) - 2. düzey. 2. >2 kap/klonda Gerçek mozaisizm - 3. düzey. Aynı bir kromozom anomalisinin; farklı kültür kaplarında ( flask tekniği) ya da farklı preparatlarda (in situ tekniği) >2 metafazda (monozomilerde >3 metafazda) saptanması ile gerçek mozaisizm tanısı konur. mos 45,X [25]/47,XXX [25]/46,XX [15] Mozaik Turner sendromu. mos 46,XY [25]/47,XY,+21 [25 ] Mozaik Down sendromu.")
15
Fetal karyotip analizlerinde mozaisizm saptanırsa??
Kadın-Doğum uzmanları ne bilmek ister? Fetus normal ? anormal ? Kötü gebelik prognozu için risk artmış mı? Kadın-Doğum uzmanları ne duymak istemezler? Yeni girişimler, ilave incelemeler, vs..

16
Örnek Olgu yanlış doğrusu
Rapordaki Bilgiler; İsim, materyalin cinsi (amniyon mayii), tarih ve “Amniyon sıvısı kültürlerinden elde metafaz plaklarının GTG bant tekniği ile yapılan mikroskobik değerlendirmesinde; 50 metafaz plağında; 38 plakta 46,XY 9 plakta 47,XXY 3 plakta 47,XY,+21 1 plakta 48,XXY,+21 kromozom kuruluşu gözlenmiştir. Gözlenen çoklu mozaik yapı nedeniyle fetusun kromozom kuruluşunun belirlenebilmesi için kord kanının değerlendirilmesi gereklidir.” yanlış doğrusu En önemli bilgi bu anomaliler kaç kültür kabında gözlendi?? Amniosentez tekrarlandı ve sonuç; fetus gerçek mozaik Klinefelter idi, trizomi 21’ li hücreler ise görülmedi ve aile gebeliğe devam kararı aldı.
, tarih ve. Amniyon sıvısı kültürlerinden elde metafaz plaklarının GTG bant tekniği ile yapılan mikroskobik değerlendirmesinde; 50 metafaz plağında; 38 plakta 46,XY. 9 plakta 47,XXY. 3 plakta 47,XY, plakta 48,XXY,+21 kromozom kuruluşu gözlenmiştir. Gözlenen çoklu mozaik yapı nedeniyle fetusun kromozom kuruluşunun belirlenebilmesi için kord kanının değerlendirilmesi gereklidir. yanlış. doğrusu. En önemli bilgi bu anomaliler kaç kültür kabında gözlendi Amniosentez tekrarlandı ve sonuç; fetus gerçek mozaik Klinefelter idi, trizomi 21’ li hücreler ise görülmedi ve aile gebeliğe devam kararı aldı.")
17
İleri anne yaşı ve patolojik USG nedeniyle uygulanan CVS de
Direkt Preparasyon 46,XX,5p+ 46,XX,5p+.ish idic(5)(p13.3)(wcp5pq++subtel5p-,5q++)
(p13.3)(wcp5pq++subtel5p-,5q++)")
18
Hücre Kültürü; 46,XX,del(5)(p13.3->pter)(subtel5p-)
Fetal karyotip; mos 46,XX,idic(5)(p13.3)/46,XX,del(5)(p13.3->pter)(subtel5p-) Yorum; Fetus 5p13.3->pter için monozomi ve 5p13.3->5qter için trizomi taşımaktadır. Mozaisizmin dokulara dağılımı kesin olarak belirlenemez. Fetusun fenotipinin etkilenmesi beklenmektedir.
(p13.3)/46,XX,del(5)(p13.3->pter)(subtel5p-) Yorum; Fetus 5p13.3->pter için monozomi ve 5p13.3->5qter için trizomi taşımaktadır. Mozaisizmin dokulara dağılımı kesin olarak belirlenemez. Fetusun fenotipinin etkilenmesi beklenmektedir.")
20
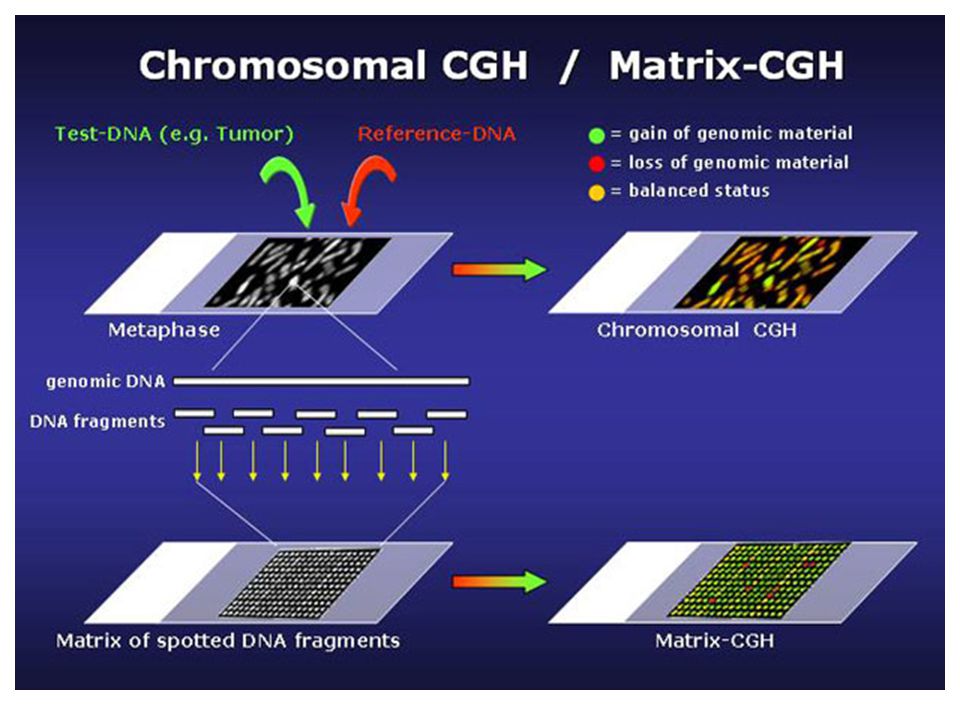
Moleküler Karyotipleme
Olgu Kontrol Cy5 Cy3

21
(7 si SNP array ile tanınabilirdi)
KARYOTİPLEME vs ARRAY Wapner R, ISCA (International Standarts for Cytogenomic Arrays) 2012 Mikroarray Çalışma Sonuçları; 4391 olgudan 51 olguda array sonuçsuz, başarı %98,8 4282 nonmozaik karyotip ve 58 mozaik karyotip Anomaliler Kromozom n Array tanıyabilir Array tanıyamaz Anöploidiler (tri 21,18,13,X,Y ve 45,X) 374 Dengesiz yapısal 22 Marker kromozom 3 2 1 (heterokromatin) Dengeli yapısal 40 Triploidi 17 (7 si SNP array ile tanınabilirdi) Toplam 456 398 (%87.3) 58 (%12.7)
 2012 Mikroarray Çalışma Sonuçları; 4391 olgudan 51 olguda array sonuçsuz, başarı %98, nonmozaik karyotip ve 58 mozaik karyotip. Anomaliler. Kromozom n. Array tanıyabilir. Array tanıyamaz. Anöploidiler (tri 21,18,13,X,Y ve 45,X) 374. Dengesiz yapısal. 22. Marker kromozom (heterokromatin) Dengeli yapısal. 40. Triploidi. 17. (7 si SNP array ile tanınabilirdi) Toplam (%87.3) 58 (%12.7)")
22
Normal karyotip saptanan prenatal olgu n:3822
Wapner R, ISCA (International Standarts for Cytogenomic Arrays) 2012 Mikroarray Çalışma Sonuçları; Normal karyotip saptanan prenatal olgu n:3822 1) Klinik anlamı bilinmeyen CNVs n: 94 (%2.5) Rapor edilmeyen CNVs n: 33 (%0.9) Rapor edilen CNVs n: 61 (%1.6) 2) Patolojik CNVs n: 35 (%0.9) Endikasyon gruplarına göre klinik ile ilişkili CNVs İleri anne yaşı n: n:34 %1.7 Positiv Tarama Testi n: n:12 %1.6 Patolojik USG n: n:45 %6 Klinik ile ilişkili CNVs %2.5
 2012 Mikroarray Çalışma Sonuçları; Normal karyotip saptanan prenatal olgu n: ) Klinik anlamı bilinmeyen CNVs n: 94 (%2.5) Rapor edilmeyen CNVs n: 33 (%0.9) Rapor edilen CNVs n: 61 (%1.6) 2) Patolojik CNVs n: 35 (%0.9) Endikasyon gruplarına göre klinik ile ilişkili CNVs. İleri anne yaşı n: 1966 n:34 %1.7. Positiv Tarama Testi n:729 n:12 %1.6. Patolojik USG n:755 n:45 %6. Klinik ile ilişkili CNVs. %2.5.")
23
Karyotipi “Normal” prenatal olgularda array sonuçları;
Schaffer LG, et al, 2012;Prenat Diagn, 32, Endikasyon Array-normal Array-şüpheli Array-patolojik Toplam olgu Pat-USG 2462 (%88.5) 135 (%4.9) 184 (%6.6) 2781 Pat-USG (soft markers) 72 (93.5) 3 (%3.9) 2 (%2.6) 77 MS-ST 68 (%88.3) 5 (%6.5) 4 (%5.2) Aile öyküsü 461 (%94.7) 11 (%2.3) 15 (%3.1) 487 İleri Anne Yaşı 337 (%97.4) 8 (%2.3) 1 (%0.3) 346 Psikolojik 94 (%98.9) 1 (%1.1) 95 Diğer/? 12 (%92.3) 1 (%7.7) 13 Toplam (canlı fetuslar 3506 (%90.5) 163 (%4.2) 207 (%5.3) 3876
 135 (%4.9) 184 (%6.6) Pat-USG (soft markers) 72 (93.5) 3 (%3.9) 2 (%2.6) 77. MS-ST. 68 (%88.3) 5 (%6.5) 4 (%5.2) Aile öyküsü. 461 (%94.7) 11 (%2.3) 15 (%3.1) 487. İleri Anne Yaşı. 337 (%97.4) 8 (%2.3) 1 (%0.3) 346. Psikolojik. 94 (%98.9) 1 (%1.1) 95. Diğer/ 12 (%92.3) 1 (%7.7) 13. Toplam (canlı fetuslar (%90.5) 163 (%4.2) 207 (%5.3)")
24
Olgu KS 112 Karyotip endikasyonu; PATUSG (ventrikulomegali, CCA, ikiz eşi) Karyotip; 47,XX,+mar a-CGH endikasyonu; marker kromozomun aydınlatılması 47,XX,+mar . arr 9p24.3-q21.11 (0-71,226,556)x4/ 9p24.3-p23(0-10,016,576)x6
x4/ 9p24.3-p23(0-10,016,576)x6.")
25
Konfirmasyon; FISH wcp 9 arm spesifik 9 p ve q subtelomerik prob 47,XX,+mar1/48,XX,+mar1,+mar2 [56/4]. ish 47,XX,+i(9)(p?)/ 48,XX,+i(9)(p?), +idic(9)(q21) 9ptel30(43N6x6, wcp9p++/wcp9q+, D9Z3++)[56/4] . arr 9p24.3-q21.11 (0-71,226,556)x4/ 9p24.3-p23(0-10,016,576)x6 Yorum; Olguda saptanan 2 marker kromozom da 9. kromozom kökenlidir. Olgu 9p24.3->q21.11 bölgesi için tetrazomi ve 9p24.3->p23 bölgesi için hekzazomi taşımaktadır. Bu anomalinin fenotipi etkilemesi beklenmektedir.
(p )/ 48,XX,+i(9)(p ), +idic(9)(q21) 9ptel30(43N6x6, wcp9p++/wcp9q+, D9Z3++)[56/4] . arr 9p24.3-q21.11 (0-71,226,556)x4/ 9p24.3-p23(0-10,016,576)x6. Yorum; Olguda saptanan 2 marker kromozom da 9. kromozom kökenlidir. Olgu 9p24.3->q21.11 bölgesi için tetrazomi ve. 9p24.3->p23 bölgesi için hekzazomi taşımaktadır. Bu anomalinin fenotipi etkilemesi beklenmektedir.")
26
Olgu GD Prenatal tanı endikasyonu; İleri anne yaşı+2’li testte artmış risk+ICSI (donuk embriodan) CVS Karyotipi 13.GH; 46,--,t(2;4)(p23;q31.1)dn a-CGH endikasyonu; görünürde dengeli de novo resiprokal translokasyon a-CGH sonucu normal 22. GH, 2. düzey USG; Ense plisi kalınlığında artış PTPN11 dizi analizi; 13. ekzonda heterozigot c.1529A>C de novo (Leopard sendromu’nda tanımlanmış bir mutasyon)
")
27
Kromozom Anomalierinin Tanısında Kullanılan Teknikler
Tüm genomu inceler Dengeli kromozom anomalilerini tanır Düşük oranlı mozaikler saptanır 3n/4n tanısı mümkün Rezolüsyon daha düşüktür (5-10 Mb) Hücre kültürü gerektirir Tanı subjektiftir Otomatizasyon zordur Lokusa /kromozoma özeldir Rezolüsyonu yüksektir <3Mb) Otomatizasyona uygundur 3n/4n tanısı mümkün Endikasyona göre hücre kültürü gerekir Endikasyona göre kullanılır Tüm genomu inceler Rezolüsyonu en yüksek (>100 Kb) Hücre kültürü gerekmez Otomatizasyona uygundur Dengeli kromozomal değişimleri tanıyamaz Düşük oranlı (<%10) mozaikleri tanıyamaz Bioinformatik çalışma gerektirir Henüz pahalı
 Hücre kültürü gerektirir. Tanı subjektiftir. Otomatizasyon zordur. Lokusa /kromozoma özeldir. Rezolüsyonu yüksektir <3Mb) Otomatizasyona uygundur. 3n/4n tanısı mümkün. Endikasyona göre hücre kültürü gerekir. Endikasyona göre kullanılır. Tüm genomu inceler. Rezolüsyonu en yüksek (>100 Kb) Hücre kültürü gerekmez. Otomatizasyona uygundur. Dengeli kromozomal değişimleri tanıyamaz. Düşük oranlı (<%10) mozaikleri tanıyamaz. Bioinformatik çalışma gerektirir. Henüz pahalı.")
28
ACOG (2009; Obstet and Gynecol, 114:5,1161-1163)
SIGU çalışma grubu (2012;Ultrasound Obstet Gynecol, 39: ) konvensiyonel karyotipin yerini almaması gerektiği, tüm gebeliklerde genel tarama için değil seçilmiş gebeliklerde özel tanı amacıyla kullanılması gerektiğini, prenatal tanıda özel endikasyonlarda; izole veya multiple USG anomalisi saptanan gebelikler ii) karyotip analizinde dengeli bile görünse de novo yapısal kromozom anomalilerinde marker kromozomların karakterizasyonunda tamamlayıcı ikinci bir test olarak kullanılmasını önermektedir “hedefe yönelik array” testi öncesinde ve sonrasında verilecek danışmalarla, anne babalar a-CGH in tüm patolojileri yakalayamayabileceği ve a-CGH sonuçlarının yorumunun zor olabileceği konusunda bilgilendirilmeli Array faydalı bir test olmakla birlikte, ilave çalışmalar (parental testler, konfirmasyonlar) gerekebilir ve maliyet artar
 konvensiyonel karyotipin yerini almaması gerektiği, tüm gebeliklerde genel tarama için değil seçilmiş gebeliklerde özel tanı amacıyla kullanılması gerektiğini, prenatal tanıda özel endikasyonlarda; izole veya multiple USG anomalisi saptanan gebelikler. ii) karyotip analizinde dengeli bile görünse de novo yapısal kromozom anomalilerinde. marker kromozomların karakterizasyonunda tamamlayıcı ikinci bir test olarak kullanılmasını önermektedir. hedefe yönelik array testi öncesinde ve sonrasında verilecek danışmalarla, anne babalar a-CGH in tüm patolojileri yakalayamayabileceği ve a-CGH sonuçlarının yorumunun zor olabileceği konusunda bilgilendirilmeli. Array faydalı bir test olmakla birlikte, ilave çalışmalar (parental testler, konfirmasyonlar) gerekebilir ve maliyet artar.")
29
ı

Benzer bir sunumlar

>")

















