Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Lipid Düşürücü İlaçlar
Lipid Düşürücü İlaçlar

2
Ders Planı Lipoproteinlerin metabolizması Antihiperlipidemik ilaçlar
HMG-KoA Redüktaz inhibitörleri (statinler) Fibratlar Nikotinik asit (niasin) Resinler Obezite tedavisinde kullanılan ilaçlar Orlistat Sibutramin
 Fibratlar. Nikotinik asit (niasin) Resinler. Obezite tedavisinde kullanılan ilaçlar. Orlistat. Sibutramin.")
3
Lipoproteinler Şilomikronlar Şilomikron Kalıntıları
Çok Düşük Dansiteli Lipoproteinler (ÇDDL) Very Low Density Lipoproteins (VLDL) Orta Dansiteli Lipoproteinler (ODL) Intermediate Density Lipoproteins (IDL) Düşük Dansiteli Lipoproteinler (DDL) Low Density Lipoproteins (LDL) Yüksek Dansiteli Lipoproteinler (YDL) High Density Lipoproteins (HDL)
 Very Low Density Lipoproteins (VLDL) Orta Dansiteli Lipoproteinler (ODL) Intermediate Density Lipoproteins (IDL) Düşük Dansiteli Lipoproteinler (DDL) Low Density Lipoproteins (LDL) Yüksek Dansiteli Lipoproteinler (YDL) High Density Lipoproteins (HDL)")
5
Çok Düşük Dansiteli Lipoprotein Yapısı
Çok Düşük Dansiteli Lipoprotein Yapısı Figure A, Although lipoproteins are similar in their basic structure, they differ in size and density. B, Very low-density lipoprotein (VLDL) structure. VLDL is a triglyceride-rich lipoprotein secreted directly by the liver. It possesses high levels of triglycerides and some esterified cholesterol in its core; its surface lipids are predominantly phosphatidylcholine and unesterified cholesterol. There are many apolipoproteins (apos) on the surface, including apos B-100, E, C-I, C-II, and C-III. The carboxy-terminal G amphipathic helix is the domain of apo C-II that activated lipoprotein lipase; lipoprotein lipase hydrolyzes triglycerides to produce VLDL remnants called intermediate-density lipoprotein (IDL, see Fig. 4-19). Further metabolism produces low-density lipoprotein (LDL) (see Fig. 4-22). During this process, excess surface remnants are removed by apo A-I-containing high-density lipoprotein (HDL) particles to produce remnant HDL particles that contain the apos C-I, C-II, and C-III. The schematic model for VLDL shown here is simplified to contain only one copy of apos C-I, C-II, C-III, and E; there may be up to 20 molecules of apo E per VLDL particle, for example. Apo B-100 is shown in a more extended conformation than that shown for LDL. (Adapted from Segrest et al. [].)
 structure. VLDL is a triglyceride-rich lipoprotein secreted directly by the liver. It possesses high levels of triglycerides and some esterified cholesterol in its core; its surface lipids are predominantly phosphatidylcholine and unesterified cholesterol. There are many apolipoproteins (apos) on the surface, including apos B-100, E, C-I, C-II, and C-III. The carboxy-terminal G amphipathic helix is the domain of apo C-II that activated lipoprotein lipase; lipoprotein lipase hydrolyzes triglycerides to produce VLDL remnants called intermediate-density lipoprotein (IDL, see Fig. 4-19). Further metabolism produces low-density lipoprotein (LDL) (see Fig. 4-22). During this process, excess surface remnants are removed by apo A-I-containing high-density lipoprotein (HDL) particles to produce remnant HDL particles that contain the apos C-I, C-II, and C-III. The schematic model for VLDL shown here is simplified to contain only one copy of apos C-I, C-II, C-III, and E; there may be up to 20 molecules of apo E per VLDL particle, for example. Apo B-100 is shown in a more extended conformation than that shown for LDL. (Adapted from Segrest et al. [].)")
6
Düşük Dansiteli Lipoprotein Yapısı
Düşük Dansiteli Lipoprotein Yapısı Figure Low-density lipoprotein (LDL) structure. This is the major cholesterol-carrying particle in humans and some other species, with a core consisting primarily of cholesteryl ester and little triglyceride and a surface of phosphatidylcholine and unesterified cholesterol. Apolipoprotein (apo) B-100, essentially the sole apo on the surface, is one of the largest proteins known with 4565 amino acid residues. Apo B serves as a ligand for the LDL receptor that mediates LDL uptake by cells. Apo B-100, shown here, is associated with the surface of LDL via two clusters of amphipathic helices located in the middle and at the carboxy-terminal end of the protein; this feature of apo B-100 structure is based on both experimental and molecular modeling studies from several laboratories. Other modeling studies suggest that the gap regions on both sides of the middle cluster of amphipathic helices are also lipid-associating but through an amphipathic β-strand motif. The amino terminal end of apo B-100 also contains amphipathic helices but they are predominantly of the non-lipid-associating G class. Apo B-100, as the result of small LDL particle size, is shown with a fully condensed structure in the β1 and β2 amphipathic β sheet domains. (Adapted from Segrest et al. [].)
 structure. This is the major cholesterol-carrying particle in humans and some other species, with a core consisting primarily of cholesteryl ester and little triglyceride and a surface of phosphatidylcholine and unesterified cholesterol. Apolipoprotein (apo) B-100, essentially the sole apo on the surface, is one of the largest proteins known with 4565 amino acid residues. Apo B serves as a ligand for the LDL receptor that mediates LDL uptake by cells. Apo B-100, shown here, is associated with the surface of LDL via two clusters of amphipathic helices located in the middle and at the carboxy-terminal end of the protein; this feature of apo B-100 structure is based on both experimental and molecular modeling studies from several laboratories. Other modeling studies suggest that the gap regions on both sides of the middle cluster of amphipathic helices are also lipid-associating but through an amphipathic β-strand motif. The amino terminal end of apo B-100 also contains amphipathic helices but they are predominantly of the non-lipid-associating G class. Apo B-100, as the result of small LDL particle size, is shown with a fully condensed structure in the β1 and β2 amphipathic β sheet domains. (Adapted from Segrest et al. [].)")
7
Yüksek Dansiteli Lipoprotein Yapısı
Yüksek Dansiteli Lipoprotein Yapısı Figure Structure of high-density lipoprotein (HDL). More than 50% of HDL weight is from apolipoproteins (apos), more than 90% of which are apos A-I and A-II. The core of HDL consists mostly of cholesteryl ester and small amounts of triglycerides, and the surface contains mostly phosphatidylcholine and unesterified cholesterol (see Fig. 4-7). Despite these common features, HDL is heterogenous in particle size or density, as well as apo composition. Not only is apo heterogeneity in the form of particles containing apo A-I (Lp A-I) and those containing both apos A-I and A-II (Lp A-I/A-II), but HDL also contains small amounts of the remainder of the exchangeable apos, apos C-I, CII, C-III, E, and A-IV. These general features are schematically illustrated here. The relative size and density of HDL can be compared with other lipoproteins (see Fig. 4-19A); the presence of multiple subpopulations of HDL is also seen. A, The Lp A-I particle. Apo C-II, C-I, and C-III are likely to be associated predominantly with this particle. B, The Lp A-I/ A-II particle is shown with apo A-II. (Adapted from Segrest et al. [].)
. More than 50% of HDL weight is from apolipoproteins (apos), more than 90% of which are apos A-I and A-II. The core of HDL consists mostly of cholesteryl ester and small amounts of triglycerides, and the surface contains mostly phosphatidylcholine and unesterified cholesterol (see Fig. 4-7). Despite these common features, HDL is heterogenous in particle size or density, as well as apo composition. Not only is apo heterogeneity in the form of particles containing apo A-I (Lp A-I) and those containing both apos A-I and A-II (Lp A-I/A-II), but HDL also contains small amounts of the remainder of the exchangeable apos, apos C-I, CII, C-III, E, and A-IV. These general features are schematically illustrated here. The relative size and density of HDL can be compared with other lipoproteins (see Fig. 4-19A); the presence of multiple subpopulations of HDL is also seen. A, The Lp A-I particle. Apo C-II, C-I, and C-III are likely to be associated predominantly with this particle. B, The Lp A-I/ A-II particle is shown with apo A-II. (Adapted from Segrest et al. [].)")
8
Triglycerides and cholesterol are transported by chylomicrons and remnant lipoproteins from the intestine and by VLDL and LDL from the liver (white arrows). ApoA-1 is synthesized by the liver and, after interaction with ABCA1, is secreted into plasma as lipid-poor apoA-1 (yellow arrow). In reverse cholesterol transport, newly synthesized lipid-poor apoA-1 interacts with ABCA1, removing excess cellular cholesterol and forming pre-beta-HDL (green arrow). Pre-beta-HDL is converted into mature alpha-HDL by LCAT (black arrow). HDL-C is returned to the liver through two pathways: selective uptake of cholesterol by the hepatic SR-B1 (blue arrow), or the transfer of cholesteryl ester by CETP to VLDL-LDL, with uptake by the liver through the LDL receptor (red arrows). Short-term HDL therapy to increase the HDL level and potentially provide protection against cardiovascular events can be achieved with the infusion of complexes consisting of apoA-1 Milano and phospholipids. Long-term increases in the HDL level and reductions in the LDL level result from the partial inhibition of CETP. HDL: high density lipoprotein; VLDL: very low density lipoprotein; LDL: low density lipoprotein; ApoA-1: apolipoprotein A-1; ABCA1: ATP-binding cassette transporter 1; LCAT: lecithin-cholesterol acyltransferase; SR-B1: scavenger receptor, class B, type I; CETP: cholesteryl ester transfer protein; FC: free cholesterol; PL: phsopholipids; LRP: LDL-related protein; LPL: lipoprotein lipase. Reproduced with permission from: Brewer, BH, Jr. Increasing HDL cholesterol levels. N Engl J Med 2004; 350:1491. Copyright ©2004 Massachusetts Medical Society. ©2006 UpToDate® • • Co
. ApoA-1 is synthesized by the liver and, after interaction with ABCA1, is secreted into plasma as lipid-poor apoA-1 (yellow arrow). In reverse cholesterol transport, newly synthesized lipid-poor apoA-1 interacts with ABCA1, removing excess cellular cholesterol and forming pre-beta-HDL (green arrow). Pre-beta-HDL is converted into mature alpha-HDL by LCAT (black arrow). HDL-C is returned to the liver through two pathways: selective uptake of cholesterol by the hepatic SR-B1 (blue arrow), or the transfer of cholesteryl ester by CETP to VLDL-LDL, with uptake by the liver through the LDL receptor (red arrows). Short-term HDL therapy to increase the HDL level and potentially provide protection against cardiovascular events can be achieved with the infusion of complexes consisting of apoA-1 Milano and phospholipids. Long-term increases in the HDL level and reductions in the LDL level result from the partial inhibition of CETP. HDL: high density lipoprotein; VLDL: very low density lipoprotein; LDL: low density lipoprotein; ApoA-1: apolipoprotein A-1; ABCA1: ATP-binding cassette transporter 1; LCAT: lecithin-cholesterol acyltransferase; SR-B1: scavenger receptor, class B, type I; CETP: cholesteryl ester transfer protein; FC: free cholesterol; PL: phsopholipids; LRP: LDL-related protein; LPL: lipoprotein lipase. Reproduced with permission from: Brewer, BH, Jr. Increasing HDL cholesterol levels. N Engl J Med 2004; 350:1491. Copyright ©2004 Massachusetts Medical Society. ©2006 UpToDate® • • Co.")
9
Lipoproteinlerin Dansiteleri ve Büyüklükleri
Lipoproteinlerin Dansiteleri ve Büyüklükleri Figure A, Although lipoproteins are similar in their basic structure, they differ in size and density. B, Very low-density lipoprotein (VLDL) structure. VLDL is a triglyceride-rich lipoprotein secreted directly by the liver. It possesses high levels of triglycerides and some esterified cholesterol in its core; its surface lipids are predominantly phosphatidylcholine and unesterified cholesterol. There are many apolipoproteins (apos) on the surface, including apos B-100, E, C-I, C-II, and C-III. The carboxy-terminal G amphipathic helix is the domain of apo C-II that activated lipoprotein lipase; lipoprotein lipase hydrolyzes triglycerides to produce VLDL remnants called intermediate-density lipoprotein (IDL, see Fig. 4-19). Further metabolism produces low-density lipoprotein (LDL) (see Fig. 4-22). During this process, excess surface remnants are removed by apo A-I-containing high-density lipoprotein (HDL) particles to produce remnant HDL particles that contain the apos C-I, C-II, and C-III. The schematic model for VLDL shown here is simplified to contain only one copy of apos C-I, C-II, C-III, and E; there may be up to 20 molecules of apo E per VLDL particle, for example. Apo B-100 is shown in a more extended conformation than that shown for LDL. (Adapted from Segrest et al. [].)
 structure. VLDL is a triglyceride-rich lipoprotein secreted directly by the liver. It possesses high levels of triglycerides and some esterified cholesterol in its core; its surface lipids are predominantly phosphatidylcholine and unesterified cholesterol. There are many apolipoproteins (apos) on the surface, including apos B-100, E, C-I, C-II, and C-III. The carboxy-terminal G amphipathic helix is the domain of apo C-II that activated lipoprotein lipase; lipoprotein lipase hydrolyzes triglycerides to produce VLDL remnants called intermediate-density lipoprotein (IDL, see Fig. 4-19). Further metabolism produces low-density lipoprotein (LDL) (see Fig. 4-22). During this process, excess surface remnants are removed by apo A-I-containing high-density lipoprotein (HDL) particles to produce remnant HDL particles that contain the apos C-I, C-II, and C-III. The schematic model for VLDL shown here is simplified to contain only one copy of apos C-I, C-II, C-III, and E; there may be up to 20 molecules of apo E per VLDL particle, for example. Apo B-100 is shown in a more extended conformation than that shown for LDL. (Adapted from Segrest et al. [].)")
10
Apoproteinler Apo Taşıyan LP tipleri İşlevi A1 YDL, şilomikronlar
Lesitin kolesterol asil transferaz (LKAT) aktivatörü A2 Hepatik lipaz aktivatörü B100 DDL, ODL, ÇDDL DDL reseptör ligandı B48 Şilomikron, şilomikron kalıntısı C1 ÇDDL, YDL LKAT aktivatörü C2 ÇDDL, YDL, şilomikronlar Ekstrahepatik lipoprotein lipaz kofaktörü C3 Ekstrahepatik lipoprotein lipaz inhibitörü D YDL (bazı alt tipleri) Kolesteril ester transfer protein kofaktörü E ÇDDL, YDL, Şilomikron, Şilomikron kalıntısı DDL reseptörü için ligand
 aktivatörü. A2. Hepatik lipaz aktivatörü. B100. DDL, ODL, ÇDDL. DDL reseptör ligandı. B48. Şilomikron, şilomikron kalıntısı. C1. ÇDDL, YDL. LKAT aktivatörü. C2. ÇDDL, YDL, şilomikronlar. Ekstrahepatik lipoprotein lipaz kofaktörü. C3. Ekstrahepatik lipoprotein lipaz inhibitörü. D. YDL (bazı alt tipleri) Kolesteril ester transfer protein kofaktörü. E. ÇDDL, YDL, Şilomikron, Şilomikron kalıntısı. DDL reseptörü için ligand.")
12
The formation and secretion of (A) chylomicrons by an intestinal cell and (B) very low density lipoproteins by a hepatic cell. (RER, rough endoplasmic reticulum; SER, smooth endoplasmic reticulum; G, Golgi apparatus; N, nucleus; C, chylomicrons; VLDL, very low density lipoproteins; E, endothelium; SD, space of Disse, containing blood plasma.) Apolipoprotein B, synthesized in the RER, is incorporated into lipoproteins in the SER, the main site of synthesis of triacylglycerol. After addition of carbohydrate residues in G, they are released from the cell by reverse pinocytosis. Chylomicrons pass into the lymphatic system. VLDL are secreted into the space of Disse and then into the hepatic sinusoids through fenestrae in the endothelial lining.
 chylomicrons by an intestinal cell and (B) very low density lipoproteins by a hepatic cell. (RER, rough endoplasmic reticulum; SER, smooth endoplasmic reticulum; G, Golgi apparatus; N, nucleus; C, chylomicrons; VLDL, very low density lipoproteins; E, endothelium; SD, space of Disse, containing blood plasma.) Apolipoprotein B, synthesized in the RER, is incorporated into lipoproteins in the SER, the main site of synthesis of triacylglycerol. After addition of carbohydrate residues in G, they are released from the cell by reverse pinocytosis. Chylomicrons pass into the lymphatic system. VLDL are secreted into the space of Disse and then into the hepatic sinusoids through fenestrae in the endothelial lining.")
13
Metabolic fate of chylomicrons. (A, apolipoprotein A; B-48, apolipoprotein B-48; , apolipoprotein C; E, apolipoprotein E; HDL, high-density lipoprotein; TG, triacylglycerol; C, cholesterol and cholesteryl ester; P, phospholipid; HL, hepatic lipase; LRP, LDL receptor-related protein.) Only the predominant lipids are shown.
 Only the predominant lipids are shown.")
15
Metabolic fate of very low density lipoproteins (VLDL) and production of low-density lipoproteins (LDL). (A, apolipoprotein A; B-100, apolipoprotein B-100; , apolipoprotein C; E, apolipoprotein E; HDL, high-density lipoprotein; TG, triacylglycerol; IDL, intermediate-density lipoprotein; C, cholesterol and cholesteryl ester; P, phospholipid.) Only the predominant lipids are shown. It is possible that some IDL is also metabolized via the LRP.
 and production of low-density lipoproteins (LDL). (A, apolipoprotein A; B-100, apolipoprotein B-100; , apolipoprotein C; E, apolipoprotein E; HDL, high-density lipoprotein; TG, triacylglycerol; IDL, intermediate-density lipoprotein; C, cholesterol and cholesteryl ester; P, phospholipid.) Only the predominant lipids are shown. It is possible that some IDL is also metabolized via the LRP.")
16
Düşük dansiteli lipoproteini hücreler nasıl tanır?
Düşük dansiteli lipoproteini hücreler nasıl tanır? Figure 9-3. A low-density lipoprotein (LDL) particle with the polar heads of lipids oriented to the outside of the sphere. Triglycerides, cholesterol, and phospholipids make up the shell. An apolipoprotein B-100 cap on the particle serves as a recognition signal for the body to use in metabolizing this particle. Dietary lipids, by influencing plasma lipids, affect the lipid composition of these particles. COOH—carboxy terminal group.
 particle with the polar heads of lipids oriented to the outside of the sphere. Triglycerides, cholesterol, and phospholipids make up the shell. An apolipoprotein B-100 cap on the particle serves as a recognition signal for the body to use in metabolizing this particle. Dietary lipids, by influencing plasma lipids, affect the lipid composition of these particles. COOH—carboxy terminal group.")
17
Figure 6-7. Receptor-mediated clearance of low-density lipoprotein (LDL). The LDL receptor is synthesized in the endoplasmic reticulum and processed in the Golgi complex. It is exported to the surface in the mature form to the plasma membrane, where it collects in the coated pits. LDL binds to its receptor in the coated pits and is internalized in the coated vesicles. After acidification and uncoating, the resulting endosomes are delivered to the lysosomes for degradation of the lipid and protein components. The receptor dissociates from the LDL and is recycled to the surface. Apo B is hydrolyzed to constituent amino acids and cholesterol esters are degraded to free cholesterol and transported to endoplasmic reticulum. This free cholesterol serves several regulatory functions. It is esterified by acyl coenzyme A:cholesterol acyl transferase (ACAT) for storage as cytoplasmic cholesteryl ester droplets. The free cholesterol suppresses activities of the key enzymes of cholesteryl biosynthetic pathway (hydroxymethyl glutaryl-coenzyme A [HMG-CoA] synthase and reductase) and suppresses the synthesis of new LDL receptor protein. (Adapted from Brown and Goldstein [].)
. The LDL receptor is synthesized in the endoplasmic reticulum and processed in the Golgi complex. It is exported to the surface in the mature form to the plasma membrane, where it collects in the coated pits. LDL binds to its receptor in the coated pits and is internalized in the coated vesicles. After acidification and uncoating, the resulting endosomes are delivered to the lysosomes for degradation of the lipid and protein components. The receptor dissociates from the LDL and is recycled to the surface. Apo B is hydrolyzed to constituent amino acids and cholesterol esters are degraded to free cholesterol and transported to endoplasmic reticulum. This free cholesterol serves several regulatory functions. It is esterified by acyl coenzyme A:cholesterol acyl transferase (ACAT) for storage as cytoplasmic cholesteryl ester droplets. The free cholesterol suppresses activities of the key enzymes of cholesteryl biosynthetic pathway (hydroxymethyl glutaryl-coenzyme A [HMG-CoA] synthase and reductase) and suppresses the synthesis of new LDL receptor protein. (Adapted from Brown and Goldstein [].)")
18
ATEROSKLEROTİK PLAK OLUŞUMU
DDL ↑ OKSİDASYON Trombosit kümelenmesi ve tromboksan salıverilmesi ↑ Monositlerin dokuya toplanması Damarda NO salıverilmesinde ↓ Makrofajlara dönüşüm Endotele bağımlı gevşeme ↓ Vazokonstriksiyon ve trombüs oluşumu Fagositoz Köpük hücresi oluşumu Damar duvarı yıkımı Trombositlerin yapışması ve sitokinlerin üretilmesi Parçalanma Oksidasyon ürünleri ve enzimlerin açığa çıkışı Epitel hasarı ATEROSKLEROTİK PLAK OLUŞUMU Düz kas proliferasyonu

19
Figure Infiltration and entrapment of low-density lipoprotein (LDL) in the arterial wall. Circulating LDLs migrate through the endothelial barrier of the arterial wall and penetrate into the intima. A portion of the LDL is entrapped in the subendothelial space as a result of its interaction with extracellular matrix components. These include the proteoglycans and other intimal glycosaminoglycans (GAGs), which have high affinity for apolipoprotein B. This entrapment increases the residence time of LDL in the artery and renders the LDL susceptible to modifications such as oxidation and aggregation. Aggregates of LDL have been identified in association with matrix components. (Adapted from Grundy [].)
 in the arterial wall. Circulating LDLs migrate through the endothelial barrier of the arterial wall and penetrate into the intima. A portion of the LDL is entrapped in the subendothelial space as a result of its interaction with extracellular matrix components. These include the proteoglycans and other intimal glycosaminoglycans (GAGs), which have high affinity for apolipoprotein B. This entrapment increases the residence time of LDL in the artery and renders the LDL susceptible to modifications such as oxidation and aggregation. Aggregates of LDL have been identified in association with matrix components. (Adapted from Grundy [].)")
20
Metabolism of high-density lipoprotein (HDL) in reverse cholesterol transport. (LCAT, lecithin:cholesterol acyltransferase; C, cholesterol; CE, cholesteryl ester; PL, phospholipid; A-I, apolipoprotein A-I; SR-B1, scavenger receptor B1; ABCA 1, ATP binding cassette transporter A1.) Pre-HDL, HDL2, HDL3—see Table 25–1. Surplus surface constituents from the action of lipoprotein lipase on chylomicrons and VLDL are another source of pre-HDL. Hepatic lipase activity is increased by androgens and decreased by estrogens, which may account for higher concentrations of plasma HDL2 in women.
 in reverse cholesterol transport. (LCAT, lecithin:cholesterol acyltransferase; C, cholesterol; CE, cholesteryl ester; PL, phospholipid; A-I, apolipoprotein A-I; SR-B1, scavenger receptor B1; ABCA 1, ATP binding cassette transporter A1.) Pre-HDL, HDL2, HDL3—see Table 25–1. Surplus surface constituents from the action of lipoprotein lipase on chylomicrons and VLDL are another source of pre-HDL. Hepatic lipase activity is increased by androgens and decreased by estrogens, which may account for higher concentrations of plasma HDL2 in women.")
21
Triacylglycerol metabolism in adipose tissue. Hormone-sensitive lipase is activated by ACTH, TSH, glucagon, epinephrine, norepinephrine, and vasopressin and inhibited by insulin, prostaglandin E1, and nicotinic acid. Details of the formation of glycerol 3-phosphate from intermediates of glycolysis are shown in Figure 24–2. (PPP, pentose phosphate pathway; TG, triacylglycerol; FFA, free fatty acids; VLDL, very low density lipoprotein.)
")
22
Control of adipose tissue lipolysis. (TSH, thyroid-stimulating hormone; FFA, free fatty acids.) Note the cascade sequence of reactions affording amplification at each step. The lipolytic stimulus is "switched off" by removal of the stimulating hormone; the action of lipase phosphatase; the inhibition of the lipase and adenylyl cyclase by high concentrations of FFA; the inhibition of adenylyl cyclase by adenosine; and the removal of cAMP by the action of phosphodiesterase. ACTH, TSH, and glucagon may not activate adenylyl cyclase in vivo, since the concentration of each hormone required in vitro is much higher than is found in the circulation. Positive () and negative () regulatory effects are represented by broken lines and substrate flow by solid lines.
 Note the cascade sequence of reactions affording amplification at each step. The lipolytic stimulus is switched off by removal of the stimulating hormone; the action of lipase phosphatase; the inhibition of the lipase and adenylyl cyclase by high concentrations of FFA; the inhibition of adenylyl cyclase by adenosine; and the removal of cAMP by the action of phosphodiesterase. ACTH, TSH, and glucagon may not activate adenylyl cyclase in vivo, since the concentration of each hormone required in vitro is much higher than is found in the circulation. Positive () and negative () regulatory effects are represented by broken lines and substrate flow by solid lines.")
23
Thermogenesis in brown adipose tissue. Activity of the respiratory chain produces heat in addition to translocating protons (Chapter 13). These protons dissipate more heat when returned to the inner mitochondrial compartment via thermogenin instead of via the F1 ATP synthase, the route that generates ATP. The passage of H+ via thermogenin is inhibited by purine nucleotides when brown adipose tissue is unstimulated. Under the influence of norepinephrine, the inhibition is removed by the production of free fatty acids (FFA) and acyl-CoA. Note the dual role of acyl-CoA in both facilitating the action of thermogenin and supplying reducing equivalents for the respiratory chain. and signify positive or negative regulatory effects.
. These protons dissipate more heat when returned to the inner mitochondrial compartment via thermogenin instead of via the F1 ATP synthase, the route that generates ATP. The passage of H+ via thermogenin is inhibited by purine nucleotides when brown adipose tissue is unstimulated. Under the influence of norepinephrine, the inhibition is removed by the production of free fatty acids (FFA) and acyl-CoA. Note the dual role of acyl-CoA in both facilitating the action of thermogenin and supplying reducing equivalents for the respiratory chain. and signify positive or negative regulatory effects.")
25
Primer Hiperlipoproteinemiler
Bozukluk Defekt Artan Lipoproteinler Klinik Kan (mg/dl) Monogenik Ailesel lipoprotein lipaz eksikliği (R) (çok nadir) Lipoprotein lipaz eksikliği Şilomikron (Tip I) X, P T:10.000 C:200 Ailesel tip-III hiperlipoproteinemi (R) (~1/10.000) Anormal apo E üretimi (apo E3 yerine apo E2) Şilomikron kalıntıları ODL (Tip III) X, A T:350 C:350 Ailesel hiperkolesterolemi (D) (~1/500) DDL reseptör eksikliği DDL (Tip IIa, seyrek olarak IIb) T:100 Ailesel hipertrigliseridemi ? ÇDDL yıkımında ↓ ya da üretiminde ↑ ÇDDL (Tip IV, seyrek olarak V) T:500 Multipl lipoprotein tip hiperlipidemi (ailesel kombine hiperlipidemi) (D) (~%1-2) DDL, ÇDDL (Tip IIa, IIb,, ya da IV, seyrek olarak V) A T: C: Multifaktoriyel Poligenik hiperkolesterolemi (~%5) DDL C:280 X: Ksantoma; P: Pankreatit; A: Prematür ateroskleroz
 Monogenik. Ailesel lipoprotein lipaz eksikliği (R) (çok nadir) Lipoprotein lipaz eksikliği. Şilomikron (Tip I) X, P. T: C:200. Ailesel tip-III hiperlipoproteinemi. (R) (~1/10.000) Anormal apo E üretimi. (apo E3 yerine apo E2) Şilomikron kalıntıları. ODL (Tip III) X, A. T:350. C:350. Ailesel hiperkolesterolemi. (D) (~1/500) DDL reseptör eksikliği. DDL (Tip IIa, seyrek olarak IIb) T:100. Ailesel hipertrigliseridemi. ÇDDL yıkımında ↓ ya da üretiminde ↑ ÇDDL (Tip IV, seyrek olarak V) T:500. Multipl lipoprotein tip hiperlipidemi (ailesel kombine hiperlipidemi) (D) (~%1-2) DDL, ÇDDL (Tip IIa, IIb,, ya da IV, seyrek olarak V) A. T: C: Multifaktoriyel. Poligenik hiperkolesterolemi (~%5) DDL. C:280. X: Ksantoma; P: Pankreatit; A: Prematür ateroskleroz.")
26
Lipoprotein paterni Plazmada yükselme Lipoprotein Lipid Tip I Şilomikron TG Tip IIa DDL K Tip IIb DDL, ÇDDL K ve TG Tip III Şilomikron kalıntıları ve ODL TG ve K Tip IV ÇDDL Tip V ÇDDL, şilomikron

27
Bazı Sekonder Hiperlipidemi Türleri
Hastalık Artan Lipoprotein Mekanizma Klinik Kan düzeyleri Diabetes mellitus ÇDDL , arasıra şilomikronlar ÇDDL sekr. , katabolizması X, P, A T: C: Hipotiroidi DDL DDL yıkımında (DDL reseptör sayısı azaldığı için) A T: C: Nefrotik sendrom ÇDDL ÇDDL ve DDL sekr. , katabolizmaları T: C: Üremi ÇDDL katabolizması T: Primer biliyer siroz kolesterol ve fosfolipid Safra kolesterol ve fosfolipidlerinin kan dolaşımına geçmesi X, A T:100 C: Alkolizm ÇDDL , genellikle şilomikronlar da ÇDDL üretimi Oral kontraseptifler (bazen şilomikronlar ) X, P X: Ksantoma; P: Pankreatit; A: Prematür ateroskleroz; T: Trigliserid (mg/dl); C: Kolesterol (mg/dl)
 A. T: C: Nefrotik sendrom. ÇDDL ÇDDL ve DDL sekr. , katabolizmaları T: C: Üremi. ÇDDL katabolizması T: Primer biliyer siroz. kolesterol. ve. fosfolipid Safra kolesterol ve. fosfolipidlerinin kan. dolaşımına geçmesi. X, A. T:100. C: Alkolizm. ÇDDL , genellikle. şilomikronlar da ÇDDL üretimi Oral kontraseptifler. (bazen şilomikronlar ) X, P. X: Ksantoma; P: Pankreatit; A: Prematür ateroskleroz; T: Trigliserid (mg/dl); C: Kolesterol (mg/dl)")
28
HMG-KoA Redüktaz İnhibitörleri (Statinler)
3-hidroksi-3-metilglutaril-koenzim A redüktaz enzimini inhibe ederler. DDL-k ~ % ↓ + resin ~ % 50 ↓ TG ~ % 25 ↓ YDL-k ~ % 10 ↑ Yan Etkileri Sık görülenler GİS bulantı, kusma, diyare Başağrısı Seyrek görülenler Myozit-benzeri sendrom Yüksek dozlarda veya CYP3A4 inhibitörleri ile birlikte alındıklarında rabdomiyoliz Hepatotoksisite Gebelerde ve emzirenlerde kontrendike

29
3-hidroksi-3-metilglutaril KoA (HMG-KoA)
Asetil KoA Asetil KoA Asetil KoA KoA Asetoasetil KoA 3-hidroksi-3-metilglutaril KoA (HMG-KoA) KoA KoA HMG-KoA redüktaz (–) Asetoasetat Asetil KoA Mevalonik asit Kolesterol Lanesterol Skualen
 KoA. KoA. HMG-KoA redüktaz. (–) Asetoasetat. Asetil KoA. Mevalonik asit. Kolesterol. Lanesterol. Skualen.")
30
Statinlerle Resin Kombinasyonunun Sinerjisi
Statinlerle Resin Kombinasyonunun Sinerjisi Figure The synergistic effects of dietary fiber and inhibitors of cholesterol biosynthesis. Dietary substances that inhibit absorption and promote excretion include fiber and phytosterols. Naturally occurring inhibitors of cholesterol biosynthesis exist in small amounts in more than 30 species of mushrooms and fungi as monacolins. Chinese red rice yeast has been shown to reduce cholesterol levels, presumably by inhibition of cholesterol biosynthesis. A, Normal metabolism; B, after bile acid sequestrants; C, after HMG-CoA reductase inhibitors. HMG CoA—hepatic hydroxymethylglutaryl coenzyme A.

33
Atorvastatin ATOR 10, 20 ve 40 mg, 30 tablet KOLESTOR 10, 20 ve 40 mg, 30 tablet LİPİTAKSİN 10, 20 ve 40 mg, 30 tablet LİPİTOR 10, 20 ve 40 mg, 30 tablet SAPHİRE 10, 20 ve 40 mg, 30 tablet TARDEN 10, 20 ve 40 mg, 30 tablet Doz: 10-20 mg/gün, P.O. Serivastatin LİPOBAY 0.2 mg ve 0.3 mg, 28 tablet Doz: mg/gün, P.O. Pravastatin PRAVACHOL 10 ve 20 mg, 20 tablet 40 mg, 30 tablet Doz: 20-40 mg/gün, P.O. Simvastatin LİPOVAS 10 mg, 28 tablet SİMVAKOL 10 mg, 28 tablet ZOCOR 10 ve 20 mg, 28 tablet 40 mg, 28 fort tablet ZOVATİN 20 ve 40 mg, 30 tablet Doz: 10-80 mg/gün, P.O. Fluvastatin LESCOL 40 mg, 28 kapsül LESCOL XL 80 mg, 28 tablet (kontrollü salıveren) Doz: 20-40 mg/gün, P.O.
 Doz: mg/gün, P.O.")
34
Fibratlar Etki mekanizmaları tam olarak aydınlatılamamıştır.
Fibratlar Etki mekanizmaları tam olarak aydınlatılamamıştır. DDL-k ~ % 10 ↓ ÇDDL YDL-k ~ % 10 ↑ gemfibrozil apoA-1 sentezini artırdığı için YDL-k ~ % ↑ Yan Etkileri Sık görülenler (%2-5) Abdominal ağrı Bulantı Diyare Seyrek görülenler Myozit-benzeri sendrom statinlerle kombinasyon, riski artırır Hepatotoksisite Raş, alopesi, impotens Safra taşı oluşumu ↑ Etki mekanizmaları: Kc’de ÇDDL sentezini ve salgılanmasını azaltırlar. Kapiler endotelinde lipoprotein lipazı stimüle ederler ÇDDL’yi azaltınca, onunla takasa giren YDL düzeyi artar (çünkü takasa girecek ÇDDL molekülleri azalmış, YDL takasa girememiş durumda kalmıştır)
 Abdominal ağrı. Bulantı. Diyare. Seyrek görülenler. Myozit-benzeri sendrom statinlerle kombinasyon, riski artırır. Hepatotoksisite. Raş, alopesi, impotens. Safra taşı oluşumu ↑ Etki mekanizmaları: Kc’de ÇDDL sentezini ve salgılanmasını azaltırlar. Kapiler endotelinde lipoprotein lipazı stimüle ederler. ÇDDL’yi azaltınca, onunla takasa giren YDL düzeyi artar (çünkü takasa girecek ÇDDL molekülleri azalmış, YDL takasa girememiş durumda kalmıştır)")
35
Fenofibrat LİPANTHYL 200 M 250 mg, 30 kapsül
Fenofibrat LİPANTHYL 200 M 250 mg, 30 kapsül LİPOFEN SR 250 mg, 30 kapsül Doz: 250 mg/gün, P.O. Gemfibrozil LOPİD 600 mg, 30 ve 100 tablet Doz: 600 mg x 2, P.O.

36
Nikotinik Asit (Niasin)
Nikotinik Asit (Niasin) Yan Etkileri Sık görülenler Flushing GİS: bulantı, kusma, diyare Seyrek görülenler Hepatit, kolestatik sarılık Hiperglisemi, hiperürisemi Kontrendikasyonlar Diyabet Peptik ülser Gut Karaciğer hastalığı olanlar Gebelik Etki mekanizmaları tam olarak aydınlatılamamıştır. ÇDDL ~ % ↓ DDL-k ~ % ↓ + resin ~ % ↓ + statin ~ % 70 ↓ YDL-k ↑ (en fazla artıran ajan) Flushing aspirin tarafından önlenir (vazodilatör prostaglandinlerin üretimine bağlıdır) Etki mekanizmaları: Kc’de ÇDDL sentezini ve salgılanmasını azaltırlar. (apo B ve ÇDDL sentezini azaltırlar) Kapiler endotelinde lipoprotein lipazı stimüle ederler Yağ dokusunda lipolizin azaltılması (intraselüler lipazın inhibisyonu) Doz 1-1.5 g x 3 kez/gün, P.O. Önce düşük doz başlanır ( mg x 3 kez) sonra giderek artırılır.
 Yan Etkileri. Sık görülenler. Flushing. GİS: bulantı, kusma, diyare. Seyrek görülenler. Hepatit, kolestatik sarılık. Hiperglisemi, hiperürisemi. Kontrendikasyonlar. Diyabet. Peptik ülser. Gut. Karaciğer hastalığı olanlar. Gebelik. Etki mekanizmaları tam olarak aydınlatılamamıştır. ÇDDL ~ % ↓ DDL-k ~ % ↓ + resin ~ % ↓ + statin ~ % 70 ↓ YDL-k ↑ (en fazla artıran ajan) Flushing aspirin tarafından önlenir (vazodilatör prostaglandinlerin üretimine bağlıdır) Etki mekanizmaları: Kc’de ÇDDL sentezini ve salgılanmasını azaltırlar. (apo B ve ÇDDL sentezini azaltırlar) Kapiler endotelinde lipoprotein lipazı stimüle ederler. Yağ dokusunda lipolizin azaltılması (intraselüler lipazın inhibisyonu) Doz g x 3 kez/gün, P.O. Önce düşük doz başlanır ( mg x 3 kez) sonra giderek artırılır.")
37
Resinler (Kolestiramin, Kolestipol)
Resinler (Kolestiramin, Kolestipol) Yan Etkileri Konstipasyon Etkileşim Tiazid diüretikler Fenilbutazon Fenobarbital Oral antikoagülanlar Digital Tetrasiklinler Demir içeren bileşikler Tiroid preparatları Barsakta safra asitleri ile kompleks yaparak onların absorbsiyonunu engellerler. DDL-k ~ % ↓ + statin ~ % ↓ TG ↑ (tedavinin başlangıcında) Kolestiramin KOLESTRAN 4 g, 30 poşet toz Doz: 4 g x 3 kez/gün, P.O. ile başlanıp g x 2-3 kez/gün dozuna kadar çıkılır
 Yan Etkileri. Konstipasyon. Etkileşim. Tiazid diüretikler. Fenilbutazon. Fenobarbital. Oral antikoagülanlar. Digital. Tetrasiklinler. Demir içeren bileşikler. Tiroid preparatları. Barsakta safra asitleri ile kompleks yaparak onların absorbsiyonunu engellerler. DDL-k ~ % ↓ + statin ~ % ↓ TG ↑ (tedavinin başlangıcında) Kolestiramin. KOLESTRAN 4 g, 30 poşet toz. Doz: 4 g x 3 kez/gün, P.O. ile başlanıp 5-10 g x 2-3 kez/gün dozuna kadar çıkılır.")
38
Çeşitli lipid düşürücülerin lipid seviyeleri üzerindeki ortalama etkileri İlaç Grubu Serum DDL-k Serum YDL-k Serum Trigliserid Resinler % % 0 veya hafif Değişiklik yok* Nikotinik asit % % % Statinler % % 5-10 % Gemfibrozil % % % Fenofibrat (mikronize form) % 6-20 % % Kolesterol absorbsiyon inhibitörleri % 17 Değişiklik yok Neomisin % * Hipertrigliseridemili hastalarda serum trigliserid düzeyleri artış göstereblilir.
 % 6-20 % % Kolesterol absorbsiyon inhibitörleri. % 17 Değişiklik yok. Neomisin. % * Hipertrigliseridemili hastalarda serum trigliserid düzeyleri artış göstereblilir.")
39
Ezetimib Kolesterolün barsaktan emilimini engeller (Putatif Transport proteini NPC1L1’i inhibe ederek). Statinlerle kombine ya da tek başına kullanılır. DDL-k ~ % ↓ (tek başına) Yan Etkileri İshal Karın ağrısı Baş ağrısı Anjiyoödem Döküntü Fibratlarla birlikte kullanılmamalıdır Goodman Gilman 2007 Ezetimibe and the Inhibition of Dietary Cholesterol Uptake Ezetimibe is the first compound approved for lowering total and LDL-C levels that inhibits cholesterol absorption by enterocytes in the small intestine (van Heek et al., 2000). It lowers LDL-C levels by about 18% and is used primarily as adjunctive therapy with statins. Outcome studies employing ezetimibe with statins are beginning, but no results are anticipated for several years (Baigent and Landry, 2003). History. Ezetimibe (SCH58235) was developed by pharmaceutical chemists studying inhibition of intestinal acyl coenzyme A:cholesterol acyltransferase (ACAT) (Burnett et al., 1994). Several compounds were found to inhibit cholesterol absorption, but by inhibiting intestinal cholesterol absorption rather than ACAT (van Heek et al., 1997). Mechanism of Action. Recent data indicate that ezetimibe inhibits a specific transport process in jejunal enterocytes, which take up cholesterol from the lumen. The putative transport protein is NPC1L1 (Altmann et al., 2004; Davis et al., 2004). In wild-type mice, ezetimibe inhibits cholesterol absorption by about 70%; in NPC1L1 knockout mice, cholesterol absorption is 86% lower than in wild-type mice, and ezetimibe has no effect on cholesterol absorption (Altmann et al., 2004). Ezetimibe does not affect intestinal triglyceride absorption. In human subjects, ezetimibe reduced cholesterol absorption by 54%, precipitating a compensatory increase in cholesterol synthesis, which can be inhibited with a cholesterol synthesis inhibitor such as a statin (Sudhop et al., 2002). There is also a substantial reduction of plasma levels of plant sterols (campesterol and sitosterol concentrations are reduced by 48% and 41%, respectively), indicating that ezetimibe also inhibits intestinal absorption of plant sterols. The consequence of inhibiting intestinal cholesterol absorption is a reduction in the incorporation of cholesterol into chylomicrons. The reduced cholesterol content of chylomicrons diminishes the delivery of cholesterol to the liver by chylomicron remnants. The diminished remnant cholesterol content may decrease atherogenesis directly, as chylomicron remnants are very atherogenic lipoproteins. In experimental animal models of remnant dyslipidemia, ezetimibe profoundly diminished diet-induced atherosclerosis (Davis et al., 2001a). Reduced delivery of intestinal cholesterol to the liver by chylomicron remnants stimulates expression of the hepatic genes regulating LDL receptor expression and cholesterol biosynthesis. The greater expression of hepatic LDL receptors enhances LDL-C clearance from the plasma. Indeed, ezetimibe reduces LDL-C levels by 15% to 20% (Gagne et al., 2002; Knopp et al., 2003). Fasting triglyceride levels decrease about 5%, and HDL-C levels increase about 1% to 2% (Dujovne et al., 2002). Combination Therapy (Ezetimibe Plus Statins). The maximal efficacy of ezetimibe for lowering LDL-C is between 15% and 20% when used as monotherapy (Gagne et al., 2002; Knopp et al., 2003). This reduction is equivalent to, or less than, that attained with 10- to 20-mg doses of most statins. Consequently, the role of ezetimibe as monotherapy of patients with elevated LDL-C levels appears to be limited to the small group of statin-intolerant patients. The actions of ezetimibe are complementary to those of statins. Statins, which inhibit cholesterol biosynthesis, increase intestinal cholesterol absorption (Miettinen and Gylling, 2003). Ezetimibe, which inhibits intestinal cholesterol absorption, enhances cholesterol biosynthesis by as much as 3.5 times in experimental animals (Davis et al., 2001b). Dual therapy with these two classes of drugs prevents the enhanced cholesterol synthesis induced by ezetimibe and the increase in cholesterol absorption induced by statins. This combination provides additive reductions in LDL-C levels irrespective of the statin employed (Ballantyne et al., 2003; Melani et al., 2003; Ballantyne et al., 2004). There is a further reduction of 15% to 20% in LDL-C when ezetimibe is combined with any statin at any dose. Increasing statin dosages from the usual starting dose of 20 mg to 80 mg normally yields only an additional 12% reduction in LDL-C, whereas adding ezetimibe, 10 mg daily, to 20 mg of a statin will reduce LDL-C by an additional 18% to 20%. A combination tablet containing ezetimibe, 10 mg, and various doses of simvastatin (10, 20, 40, and 80 mg) has been approved (VYTORIN). At the highest simvastatin dose (80 mg), plus ezetimibe (10 mg), average LDL-C reduction was 60%, which is greater than can be attained with any statin as monotherapy (Feldman et al., 2004). Absorption, Fate, and Excretion. Ezetimibe is highly water insoluble, precluding studies of its bioavailability. After ingestion, it is glucuronidated in the intestinal epithelium, absorbed, and enters an enterohepatic recirculation (Patrick et al., 2002). Pharmacokinetic studies indicate that about 70% is excreted in the feces and about 10% in the urine (as a glucuronide conjugate) (Patrick et al., 2002). Bile acid sequestrants inhibit absorption of ezetimibe, and the two agents should not be administered together. Otherwise, no significant drug interactions have been reported. Adverse Effects and Drug Interactions. Other than rare allergic reactions, specific adverse effects have not been observed in patients taking ezetimibe. The safety of ezetimibe during pregnancy has not been established. With doses of ezetimibe sufficient to increase exposure 10 to 150 times compared with a 10-mg dose in humans, fetal skeletal abnormalities were noted in rats and rabbits. Since all statins are contraindicated in pregnant and nursing women, combination products containing ezetimibe and a statin should not be used by women in childbearing years in the absence of contraception. Therapeutic Uses. Ezetimibe (ZETIA) is available as a 10-mg tablet that may be taken at any time during the day, with or without food. Ezetimibe may be taken with any medication other than bile acid sequestrants, which inhibit its absorption. EZETROL 10 mg, 7 tablet/kutu ve 28 tablet/kutu Doz: 10 mg/gün, P.O.
 Yan Etkileri. İshal. Karın ağrısı. Baş ağrısı. Anjiyoödem. Döküntü. Fibratlarla birlikte kullanılmamalıdır. Goodman Gilman Ezetimibe and the Inhibition of Dietary Cholesterol Uptake Ezetimibe is the first compound approved for lowering total and LDL-C levels that inhibits cholesterol absorption by enterocytes in the small intestine (van Heek et al., 2000). It lowers LDL-C levels by about 18% and is used primarily as adjunctive therapy with statins. Outcome studies employing ezetimibe with statins are beginning, but no results are anticipated for several years (Baigent and Landry, 2003). History. Ezetimibe (SCH58235) was developed by pharmaceutical chemists studying inhibition of intestinal acyl coenzyme A:cholesterol acyltransferase (ACAT) (Burnett et al., 1994). Several compounds were found to inhibit cholesterol absorption, but by inhibiting intestinal cholesterol absorption rather than ACAT (van Heek et al., 1997). Mechanism of Action. Recent data indicate that ezetimibe inhibits a specific transport process in jejunal enterocytes, which take up cholesterol from the lumen. The putative transport protein is NPC1L1 (Altmann et al., 2004; Davis et al., 2004). In wild-type mice, ezetimibe inhibits cholesterol absorption by about 70%; in NPC1L1 knockout mice, cholesterol absorption is 86% lower than in wild-type mice, and ezetimibe has no effect on cholesterol absorption (Altmann et al., 2004). Ezetimibe does not affect intestinal triglyceride absorption. In human subjects, ezetimibe reduced cholesterol absorption by 54%, precipitating a compensatory increase in cholesterol synthesis, which can be inhibited with a cholesterol synthesis inhibitor such as a statin (Sudhop et al., 2002). There is also a substantial reduction of plasma levels of plant sterols (campesterol and sitosterol concentrations are reduced by 48% and 41%, respectively), indicating that ezetimibe also inhibits intestinal absorption of plant sterols. The consequence of inhibiting intestinal cholesterol absorption is a reduction in the incorporation of cholesterol into chylomicrons. The reduced cholesterol content of chylomicrons diminishes the delivery of cholesterol to the liver by chylomicron remnants. The diminished remnant cholesterol content may decrease atherogenesis directly, as chylomicron remnants are very atherogenic lipoproteins. In experimental animal models of remnant dyslipidemia, ezetimibe profoundly diminished diet-induced atherosclerosis (Davis et al., 2001a). Reduced delivery of intestinal cholesterol to the liver by chylomicron remnants stimulates expression of the hepatic genes regulating LDL receptor expression and cholesterol biosynthesis. The greater expression of hepatic LDL receptors enhances LDL-C clearance from the plasma. Indeed, ezetimibe reduces LDL-C levels by 15% to 20% (Gagne et al., 2002; Knopp et al., 2003). Fasting triglyceride levels decrease about 5%, and HDL-C levels increase about 1% to 2% (Dujovne et al., 2002). Combination Therapy (Ezetimibe Plus Statins). The maximal efficacy of ezetimibe for lowering LDL-C is between 15% and 20% when used as monotherapy (Gagne et al., 2002; Knopp et al., 2003). This reduction is equivalent to, or less than, that attained with 10- to 20-mg doses of most statins. Consequently, the role of ezetimibe as monotherapy of patients with elevated LDL-C levels appears to be limited to the small group of statin-intolerant patients. The actions of ezetimibe are complementary to those of statins. Statins, which inhibit cholesterol biosynthesis, increase intestinal cholesterol absorption (Miettinen and Gylling, 2003). Ezetimibe, which inhibits intestinal cholesterol absorption, enhances cholesterol biosynthesis by as much as 3.5 times in experimental animals (Davis et al., 2001b). Dual therapy with these two classes of drugs prevents the enhanced cholesterol synthesis induced by ezetimibe and the increase in cholesterol absorption induced by statins. This combination provides additive reductions in LDL-C levels irrespective of the statin employed (Ballantyne et al., 2003; Melani et al., 2003; Ballantyne et al., 2004). There is a further reduction of 15% to 20% in LDL-C when ezetimibe is combined with any statin at any dose. Increasing statin dosages from the usual starting dose of 20 mg to 80 mg normally yields only an additional 12% reduction in LDL-C, whereas adding ezetimibe, 10 mg daily, to 20 mg of a statin will reduce LDL-C by an additional 18% to 20%. A combination tablet containing ezetimibe, 10 mg, and various doses of simvastatin (10, 20, 40, and 80 mg) has been approved (VYTORIN). At the highest simvastatin dose (80 mg), plus ezetimibe (10 mg), average LDL-C reduction was 60%, which is greater than can be attained with any statin as monotherapy (Feldman et al., 2004). Absorption, Fate, and Excretion. Ezetimibe is highly water insoluble, precluding studies of its bioavailability. After ingestion, it is glucuronidated in the intestinal epithelium, absorbed, and enters an enterohepatic recirculation (Patrick et al., 2002). Pharmacokinetic studies indicate that about 70% is excreted in the feces and about 10% in the urine (as a glucuronide conjugate) (Patrick et al., 2002). Bile acid sequestrants inhibit absorption of ezetimibe, and the two agents should not be administered together. Otherwise, no significant drug interactions have been reported. Adverse Effects and Drug Interactions. Other than rare allergic reactions, specific adverse effects have not been observed in patients taking ezetimibe. The safety of ezetimibe during pregnancy has not been established. With doses of ezetimibe sufficient to increase exposure 10 to 150 times compared with a 10-mg dose in humans, fetal skeletal abnormalities were noted in rats and rabbits. Since all statins are contraindicated in pregnant and nursing women, combination products containing ezetimibe and a statin should not be used by women in childbearing years in the absence of contraception. Therapeutic Uses. Ezetimibe (ZETIA) is available as a 10-mg tablet that may be taken at any time during the day, with or without food. Ezetimibe may be taken with any medication other than bile acid sequestrants, which inhibit its absorption. EZETROL 10 mg, 7 tablet/kutu ve 28 tablet/kutu. Doz: 10 mg/gün, P.O.")
40
Is combined treatment with fenofibrate and ezetimibe safe and efficacious in patients with mixed hyperlipidemia? Michael H Davidson Correspondence Radiant Research, 515 North State Street, Suite 2700, Chicago, IL 60610, USA This article has no abstract so we have provided the first paragraph of the full text. Ezetimibe, a novel cholesterol absorption inhibitor, has become a well-recognized therapy for lowering LDL cholesterol, most commonly in combination with a statin. By lowering LDL cholesterol by a further 18% over statin alone, ezetimibe treatment results in LDL cholesterol reductions comparable to that of triple the statin dose and has, therefore, become a very useful treatment to achieve National Cholesterol Education Program Adult Treatment Panel III goals in high-risk patients. When ezetimibe first became available, the combination with fibrate was not advised because of the lack of data from large safety and efficacy trials, and the evidence from pharmacokinetic trials demonstrating that fibrates increased the bioavailability of ezetimibe, resulting in higher plasma levels.1 In addition, fibrates are known to increase cholelithiasis, and a dog model found that ezetimibe increased the cholesterol content of bile, thereby theoretically increasing the risk of gallstones.2 Higher blood levels of ezetimibe were not believed to be a safety issue because in dose-ranging trials doses above 10 mg were not associated with additional adverse effects. An initial 12-week placebo-controlled trial3 comparing ezetimibe and fenofibrate combined or alone demonstrated the added efficacy of coadministered ezetimibe and fenofibrate compared with the monotherapy of each treatment, but since the trial only lasted 12 weeks, long-term safety, especially in reference to liver abnormalities or gallbladder disease, could not be established. This 48-week trial by McKenney et al. has confirmed the sustained efficacy of the combined therapy shown in the 12-week trial and established that there are no significant adverse effects associated with a combination of ezetimibe and a fibrate compared with fibrate alone. Is combined treatment with fenofibrate and ezetimibe safe and efficacious in patients with mixed hyperlipidemia? Michael H Davidson Correspondence Radiant Research, 515 North State Street, Suite 2700, Chicago, IL 60610, USA This article has no abstract so we have provided the first paragraph of the full text. Ezetimibe, a novel cholesterol absorption inhibitor, has become a well-recognized therapy for lowering LDL cholesterol, most commonly in combination with a statin. By lowering LDL cholesterol by a further 18% over statin alone, ezetimibe treatment results in LDL cholesterol reductions comparable to that of triple the statin dose and has, therefore, become a very useful treatment to achieve National Cholesterol Education Program Adult Treatment Panel III goals in high-risk patients. When ezetimibe first became available, the combination with fibrate was not advised because of the lack of data from large safety and efficacy trials, and the evidence from pharmacokinetic trials demonstrating that fibrates increased the bioavailability of ezetimibe, resulting in higher plasma levels.1 In addition, fibrates are known to increase cholelithiasis, and a dog model found that ezetimibe increased the cholesterol content of bile, thereby theoretically increasing the risk of gallstones.2 Higher blood levels of ezetimibe were not believed to be a safety issue because in dose-ranging trials doses above 10 mg were not associated with additional adverse effects. An initial 12-week placebo-controlled trial3 comparing ezetimibe and fenofibrate combined or alone demonstrated the added efficacy of coadministered ezetimibe and fenofibrate compared with the monotherapy of each treatment, but since the trial only lasted 12 weeks, long-term safety, especially in reference to liver abnormalities or gallbladder disease, could not be established. This 48-week trial by McKenney et al. has confirmed the sustained efficacy of the combined therapy shown in the 12-week trial and established that there are no significant adverse effects associated with a combination of ezetimibe and a fibrate compared with fibrate alone.

41
Inhibitors of Cholesteryl Ester Transfer Protein
Inhibitors of Cholesteryl Ester Transfer Protein The cholesteryl ester transfer protein (CETP) is a plasma glycoprotein synthesized by the liver that mediates the transfer of cholesteryl esters from the larger subfractions of HDL (HDL2) to triglyceride-rich lipoproteins and LDL in exchange for a molecule of triglyceride. Enrichment of HDL2 with triglycerides enhances its catabolism by the liver. In animal models, inhibition of CETP results in higher HDL levels, decreased LDL levels, and resistance to developing atherosclerosis. Observational studies of humans with CETP gene mutations associated with reduced CETP activity indicate that HDL levels are increased and LDL levels are lower in affected patients. However, there are reports of both increased and decreased prevalence of CHD, or no effect on CHD prevalence in patients with naturally occurring CETP mutations. Clinical trials of CETP inhibitors in human subjects are under way (see review by Forrester et al., 2005). Two CETP inhibitors, JTT-705 and torcetrapib, are being tested (de Grooth et al., 2002; Brousseau et al., 2004; Clark et al., 2004). JTT-705 forms a disulfide bond with CETP, and torcetrapib is thought to stabilize the association of CETP with its lipoprotein substrate, creating a nonfunctional complex. The levels of HDL-C are increased by 45% to 106% in normal subjects and in patients with low HDL-C levels. Further studies of the safety of these compounds and proof that this approach prevents clinical vascular disease are required before CETP inhibitors can be routinely used in managing dyslipidemic patients.
 is a plasma glycoprotein synthesized by the liver that mediates the transfer of cholesteryl esters from the larger subfractions of HDL (HDL2) to triglyceride-rich lipoproteins and LDL in exchange for a molecule of triglyceride. Enrichment of HDL2 with triglycerides enhances its catabolism by the liver. In animal models, inhibition of CETP results in higher HDL levels, decreased LDL levels, and resistance to developing atherosclerosis. Observational studies of humans with CETP gene mutations associated with reduced CETP activity indicate that HDL levels are increased and LDL levels are lower in affected patients. However, there are reports of both increased and decreased prevalence of CHD, or no effect on CHD prevalence in patients with naturally occurring CETP mutations. Clinical trials of CETP inhibitors in human subjects are under way (see review by Forrester et al., 2005). Two CETP inhibitors, JTT-705 and torcetrapib, are being tested (de Grooth et al., 2002; Brousseau et al., 2004; Clark et al., 2004). JTT-705 forms a disulfide bond with CETP, and torcetrapib is thought to stabilize the association of CETP with its lipoprotein substrate, creating a nonfunctional complex. The levels of HDL-C are increased by 45% to 106% in normal subjects and in patients with low HDL-C levels. Further studies of the safety of these compounds and proof that this approach prevents clinical vascular disease are required before CETP inhibitors can be routinely used in managing dyslipidemic patients.")
42
Obezite Tedavisinde Kullanılan İlaçlar Orlistat
Obezite Tedavisinde Kullanılan İlaçlar Orlistat Gastrik ve pankreatik lipazı inhibe ederek diyetle alınan yağların emilmesini azaltır. 2 yıldan uzun süre kullanılmamalıdır. Yan Etkileri Steatore Flatulans Orlistat XENİCAL 120 mg, 84 kapsül/kutu Doz: 120 mg x 3 kez/gün, PO

43
Figure Effects of orlistat over a 2-year period [][]. Inhibition of lipases by orlistat blocks systemic absorption of dietary fat. Up to one third of ingested fat is excreted into the feces. Gastric and pancreatic lipases are the key enzymes that hydrolyze triglycerides into free fatty acids and monoglycerides, which are then absorbed. Orlistat inhibits these lipases.

44
Obezite tedavisinde Kullanılan İlaçlar Sibutramin
Obezite tedavisinde Kullanılan İlaçlar Sibutramin Noradrenalin ve serotonin reuptake blokörüdür. İştahı azaltıcı etki gösterir. 1 yıldan uzun süre kullanılmamalıdır. Yan Etkileri (seyrek) Konstipasyon Anoreksi Ağız kuruluğu Uykusuzluk Çarpıntı Hipertansiyon Anksiyete Tad duyusunda bozulma REDUCTİL 10 ve 15 mg, 28 kapsül/kutu ZELIUM 10 ve 15 mg, 28 kapsül/kutu Doz: 10 mg (sabah) PO ile başlanır.
 Konstipasyon. Anoreksi. Ağız kuruluğu. Uykusuzluk. Çarpıntı. Hipertansiyon. Anksiyete. Tad duyusunda bozulma. REDUCTİL 10 ve 15 mg, 28 kapsül/kutu. ZELIUM 10 ve 15 mg, 28 kapsül/kutu. Doz: 10 mg (sabah) PO ile başlanır.")
45
Ağır hipertrigliseridemi tedavisinde öncelikle tercih edilmesi gereken ilaç aşağıdakilerden hangisidir? A) Nikotinik asit B) Gemfibrozil C) Atorvastatin D) Simvastatin E) Kolastiramin (Cevap B) 2004 Nisan
 Nikotinik asit. B) Gemfibrozil. C) Atorvastatin. D) Simvastatin. E) Kolastiramin. (Cevap B) 2004 Nisan.")
46
Figure The major pathways involved in the metabolism of chylomicrons synthesized by the intestine and VLDL synthesized by the liver. Chylomicrons are converted to chylomicron remnants by the hydrolysis of their triglycerides by LPL. Chylomicron remnants are rapidly cleared from the plasma by the liver. "Remnant receptors" include the LDL receptor-related protein (LRP), LDL, and perhaps other receptors. FFA released by LPL is used by muscle tissue as an energy source or taken up and stored by adipose tissue. FFA, free fatty acid; HL, hepatic lipase; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins.
, LDL, and perhaps other receptors. FFA released by LPL is used by muscle tissue as an energy source or taken up and stored by adipose tissue. FFA, free fatty acid; HL, hepatic lipase; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins.")
49
Drug Effects on HDL: Niacin
Drug Effects on HDL: Niacin B TG C-II NIACIN LPL CM/VLDL Intestine B Drug Effects on HDL: Niacin This slide illustrates the possible mechanism by which niacin increases high-density lipoprotein (HDL) levels. Niacin decreases very-low-density lipoprotein (VLDL) synthesis by the liver, which results in increased levels of HDL. By selectively decreasing triglyceride (TG) synthesis, through the inhibition of both the synthesis and esterification of fatty acids, niacin accelerates hepatic intracellular post-translational degradation of apolipoprotein (apo) B. Niacin also selectively decreases hepatic removal of apo A-I (but not cholesterol ester) from HDL. References: Jin F-Y, Kamanna VS, Kashyap ML. Niacin accelerates intracellular apoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arterioscler Thromb Vasc Biol. 1999;19:1051–1059. Jin F-Y, Kamanna VS, Kashyap ML. Niacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells: implication for reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 1997;17:2020–2028. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR CMR/IDL NIACIN A-I A-I * CE LCAT FC FC CE ABC1 Liver Nascent HDL HL Macrophage Mature HDL *Inhibits uptake of apoA-I but not CE
 levels. Niacin decreases very-low-density lipoprotein (VLDL) synthesis by the liver, which results in increased levels of HDL. By selectively decreasing triglyceride (TG) synthesis, through the inhibition of both the synthesis and esterification of fatty acids, niacin accelerates hepatic intracellular post-translational degradation of apolipoprotein (apo) B. Niacin also selectively decreases hepatic removal of apo A-I (but not cholesterol ester) from HDL. References: Jin F-Y, Kamanna VS, Kashyap ML. Niacin accelerates intracellular apoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arterioscler Thromb Vasc Biol. 1999;19:1051–1059. Jin F-Y, Kamanna VS, Kashyap ML. Niacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells: implication for reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 1997;17:2020–2028. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR. CMR/IDL. NIACIN. A-I. A-I. * CE. LCAT. FC. FC. CE. ABC1. Liver. Nascent HDL. HL. Macrophage. Mature HDL. *Inhibits uptake of apoA-I but not CE.")
50
Drug Effects on HDL: Fibrates
Drug Effects on HDL: Fibrates B FIBRATES + TG C-II LPL CM/VLDL Intestine B Drug Effects on HDL: Fibrates This slide illustrates the mechanisms by which fibrates increase high-density lipoprotein (HDL) levels. The mechanisms of action of fibrates are only partially understood, but they appear to activate specific transcription factors belonging to the nuclear hormone receptor superfamily, the peroxisome proliferator-activated receptors (PPARs). PPAR- mediates fibrate action on HDL-cholesterol levels via transcriptional induction of synthesis of the major HDL apolipoproteins (apoA-I and apoA-II) as well as lipoprotein lipase. Fibrates decrease hepatic apoC-III transcription, thereby enhancing clearance of triglyceride-rich lipoproteins. Reference: Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–2093. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR + CMR/IDL A-I FIBRATES A-I CE LCAT FC FC CE ABC1 Liver Nascent HDL HL Macrophage Mature HDL
 levels. The mechanisms of action of fibrates are only partially understood, but they appear to activate specific transcription factors belonging to the nuclear hormone receptor superfamily, the peroxisome proliferator-activated receptors (PPARs). PPAR- mediates fibrate action on HDL-cholesterol levels via transcriptional induction of synthesis of the major HDL apolipoproteins (apoA-I and apoA-II) as well as lipoprotein lipase. Fibrates decrease hepatic apoC-III transcription, thereby enhancing clearance of triglyceride-rich lipoproteins. Reference: Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–2093. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR. + CMR/IDL. A-I. FIBRATES. A-I. CE. LCAT. FC. FC. CE. ABC1. Liver. Nascent HDL. HL. Macrophage. Mature HDL.")
51
Drug Effects on HDL: Estrogens
Drug Effects on HDL: Estrogens B TG C-II LPL CM/VLDL Intestine B Drug Effects on HDL: Estrogens This slide illustrates the mechanisms by which estrogens increase high-density lipoprotein (HDL) levels. Estrogens increase apolipoprotein A-I production and inhibit the activity of hepatic lipase, leading to increased production of mature HDL. Estrogens also may induce hypertriglyceridemia by increasing the rate of very-low-density lipoprotein (VLDL) production. Furthermore, low-density lipoprotein (LDL) levels may decrease with estrogen use because of increased LDL receptors and increased LDL removal in the liver. References: Walsh BW, Schiff I, Rosner B, et al. Effects of postmenopausal estrogen replacement on the concentrations and metabolism of plasma lipoproteins. N Engl J Med. 1991;325:1196–1204. Miller KW, Lane MD. Estradiol-induced alteration of very-low-density lipoprotein assembly. J Biol Chem. 1984;259:15277–15286. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR + CMR/IDL A-I ESTROGENS A-I CE LCAT FC FC CE ABC1 Liver Nascent HDL HL Macrophage Mature HDL ESTROGENS
 levels. Estrogens increase apolipoprotein A-I production and inhibit the activity of hepatic lipase, leading to increased production of mature HDL. Estrogens also may induce hypertriglyceridemia by increasing the rate of very-low-density lipoprotein (VLDL) production. Furthermore, low-density lipoprotein (LDL) levels may decrease with estrogen use because of increased LDL receptors and increased LDL removal in the liver. References: Walsh BW, Schiff I, Rosner B, et al. Effects of postmenopausal estrogen replacement on the concentrations and metabolism of plasma lipoproteins. N Engl J Med. 1991;325:1196–1204. Miller KW, Lane MD. Estradiol-induced alteration of very-low-density lipoprotein assembly. J Biol Chem. 1984;259:15277– Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR. + CMR/IDL. A-I. ESTROGENS. A-I. CE. LCAT. FC. FC. CE. ABC1. Liver. Nascent HDL. HL. Macrophage. Mature HDL. ESTROGENS.")
52
Drug Effects on HDL: Statins
Drug Effects on HDL: Statins B TG C-II LPL CM/VLDL + Intestine B Drug Effects on HDL: Statins This slide illustrates the mechanism by which statins affect high-density lipoprotein (HDL) metabolism. All statins are inhibitors of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting step in cholesterol biosynthesis. The predominant effect of statins is to increase low-density lipoprotein (LDL) clearance, thus reducing LDL cholesterol levels. Only recently have studies begun to examine the effects of statins on HDL cholesterol (HDL-C) levels and HDL metabolism and to determine whether pharmacologic differences among statins lead to differing effects on HDL-C levels. Statins not only inhibit cholesterol synthesis but are also believed to increase apolipoprotein A-I production, and may inhibit the activity of hepatic lipase, leading to increased production of mature HDL. Reference: Schaefer JR, Schweer H, Ikewaki H, et al. Metabolic basis of high density lipoprotein and apolipoprotein A-I increase by HMG-CoA reductase inhibition in healthy subjects and a patient with coronary artery disease. Atherosclerosis. 1999;144:177–184. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR STATINS + CMR/IDL A-I STATINS A-I CE LCAT FC FC CE ABC1 Liver Nascent HDL HL ? Macrophage Mature HDL STATINS
 metabolism. All statins are inhibitors of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting step in cholesterol biosynthesis. The predominant effect of statins is to increase low-density lipoprotein (LDL) clearance, thus reducing LDL cholesterol levels. Only recently have studies begun to examine the effects of statins on HDL cholesterol (HDL-C) levels and HDL metabolism and to determine whether pharmacologic differences among statins lead to differing effects on HDL-C levels. Statins not only inhibit cholesterol synthesis but are also believed to increase apolipoprotein A-I production, and may inhibit the activity of hepatic lipase, leading to increased production of mature HDL. Reference: Schaefer JR, Schweer H, Ikewaki H, et al. Metabolic basis of high density lipoprotein and apolipoprotein A-I increase by HMG-CoA reductase inhibition in healthy subjects and a patient with coronary artery disease. Atherosclerosis. 1999;144:177–184. Acronyms used in slide: A-I = apolipoprotein A-I; ABC1 = ATP-binding cassette protein 1; B = apolipoprotein B; C-II = apolipoprotein C-II; CE = cholesteryl ester; CM = chylomicron; CMR = chylomicron remnant; FC = free cholesterol; HDL = high-density lipoprotein; HL = hepatic lipase; IDL = intermediate-density lipoprotein; LCAT = lecithin:cholesterol acyltransferase; LDLR = low-density lipoprotein receptor; LPL = lipoprotein lipase; TG = triglyceride. LDLR. STATINS. + CMR/IDL. A-I. STATINS. A-I. CE. LCAT. FC. FC. CE. ABC1. Liver. Nascent HDL. HL. Macrophage. Mature HDL. STATINS.")
53
Possible Atherogenic Changes Accompanying Hypertriglyceridemia
Possible Atherogenic Changes Accompanying Hypertriglyceridemia Increased VLDL cholesterol-rich remnants Small, dense LDL Low HDL Plasma triglyceride levels less than 200 mg/dL are classified as normal. There has been some debate about whether an elevated triglyceride level is an independent risk factor for atherosclerosis. However, it is clearly associated with a number of metabolic or physiologic changes that are risk factors for this disease. These include: Low HDL Unusually atherogenic forms of LDL Elevated triglycerides often reflect an increase in triglyceride-rich remnant lipoproteins that have atherogenic potential. Expert Panel. JAMA. 1993;269: Hypertriglyceridemia Coagulation changes Increased chylomicron remnants Miller M. Eur Heart J. 1998;19(Suppl H):H18-H22.
:H18-H22.")
54
Atherogenicity of Small, Dense LDL
Atherogenicity of Small, Dense LDL Endothelial Chemoattractants LDL Endothelium Monocyte LDL Macrophage Small, dense LDL binds with poor affinity to the LDL receptor and, therefore, remains in plasma for prolonged periods. Macrophage uptake of small, dense LDL particles may result from the action of oxygen free radicals on these particles. Small, dense LDL particles possess low degrees of oxidative resistance and are more susceptible to oxidative modification. As a result, cholesterol-rich, macrophage foam cells may form. Macrophage foam cells are characteristic of lipid-rich plaques. They also secrete many factors that contribute to the proatherogenic, proinflammatory, and prothrombogenic activities. Chapman MJ, et al. Eur Heart J. 1998;19(suppl A):A24-A30. Mildly oxidized Macrophage Foam Cell Smooth Muscle Cell Highly oxidized Evidence from in vitro studies suggests that large, buoyant LDL particles are more resistant to oxidative stress and small, dense LDL particles more susceptible to oxidation.
:A24-A30. Mildly oxidized. Macrophage. Foam Cell. Smooth Muscle Cell. Highly oxidized. Evidence from in vitro studies suggests that large, buoyant LDL particles are more resistant to oxidative stress and small, dense LDL particles more susceptible to oxidation.")
55
Lipoprotein Classes and Inflammation
Lipoprotein Classes and Inflammation Chylomicrons, VLDL, and their catabolic remnants LDL HDL Lipoprotein classes and inflammation All the major lipoprotein classes impact in some way on the inflammatory process that leads to development of atherosclerosis. The triglyceride-rich lipoproteins—chylomicrons, very low density lipoprotein (VLDL), and their catabolic remnants—and low-density lipoprotein (LDL) are potentially proinflammatory, whereas high-density lipoprotein (HDL) is potentially anti-inflammatory. LDL Of all of the plasma lipoproteins, LDL has been most investigated in terms of its role in inflammation. LDL consists of a surface monolayer of phospholipids and free cholesterol and a single molecule of apolipoprotein (apo) B, which encircles the lipoprotein. This surface monolayer surrounds a hydrophobic core of mainly cholesteryl esters but also some triglycerides. In itself, LDL is almost certainly not proinflammatory, but the particle can become modified in many ways. It is the modified LDL particle that is proinflammatory and proatherogenic. Structure of HDL HDL has the same essential structure as LDL, with a surface monolayer of phospholipids and free cholesterol and a hydrophobic core consisting mainly of cholesteryl esters but also some triglyceride. However, HDL particles are smaller and contain different apolipoproteins, mainly apo A-I and apo A-II. Both these apolipoproteins have properties that protect the lipids against oxidative modification. In addition, some of the other proteins transported by HDL, such as paraoxonase, have antioxidant properties. Therefore, whereas LDL is very susceptible to oxidative modification, HDL is relatively resistant to it, and this is one of the reasons underlying the anti-inflammatory properties of HDL. apoB apoE apoA-I apoA-II apoE > 30 nm 20–22 nm 9–15 nm Potentially anti- inflammatory Potentially proinflammatory Doi H et al. Circulation 2000;102: ; Colome C et al. Atherosclerosis 2000; 149: ; Cockerill GW et al. Arterioscler Thromb Vasc Biol 1995;15: Slide Source: Lipids Online
, and their catabolic remnants—and low-density lipoprotein (LDL) are potentially proinflammatory, whereas high-density lipoprotein (HDL) is potentially anti-inflammatory. LDL. Of all of the plasma lipoproteins, LDL has been most investigated in terms of its role in inflammation. LDL consists of a surface monolayer of phospholipids and free cholesterol and a single molecule of apolipoprotein (apo) B, which encircles the lipoprotein. This surface monolayer surrounds a hydrophobic core of mainly cholesteryl esters but also some triglycerides. In itself, LDL is almost certainly not proinflammatory, but the particle can become modified in many ways. It is the modified LDL particle that is proinflammatory and proatherogenic. Structure of HDL. HDL has the same essential structure as LDL, with a surface monolayer of phospholipids and free cholesterol and a hydrophobic core consisting mainly of cholesteryl esters but also some triglyceride. However, HDL particles are smaller and contain different apolipoproteins, mainly apo A-I and apo A-II. Both these apolipoproteins have properties that protect the lipids against oxidative modification. In addition, some of the other proteins transported by HDL, such as paraoxonase, have antioxidant properties. Therefore, whereas LDL is very susceptible to oxidative modification, HDL is relatively resistant to it, and this is one of the reasons underlying the anti-inflammatory properties of HDL. apoB. apoE. apoA-I. apoA-II. apoE. > 30 nm. 20–22 nm. 9–15 nm. Potentially anti- inflammatory. Potentially proinflammatory. Doi H et al. Circulation 2000;102: ; Colome C et al. Atherosclerosis 2000; 149: ; Cockerill GW et al. Arterioscler Thromb Vasc Biol 1995;15: Slide Source: Lipids Online.")
56
Role of LDL in Inflammation
Role of LDL in Inflammation LDL Readily Enter the Artery Wall Where They May be Modified LDL Vessel Lumen Endothelium Role of LDL in inflammation LDL readily enters the artery wall by crossing the endothelial membrane. Once in the arterial wall, if LDL accumulates, it is subject to a variety of modifications. The best known of these is oxidation, both of the lipids and of the apo B. LDL is also subject to aggregation, and its phospholipids are subject to hydrolysis by phospholipases to form lysophosphatidylcholine. Several other chemical modifications have also been reported. The net effect of these changes is the production of a variety of modified LDL particles, and the evidence is now very strong that these modified LDL particles are proinflammatory. Reference: Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol: modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 1989;320: LDL Oxidation of Lipids and ApoB Hydrolysis of Phosphatidylcholine to Lysophosphatidylcholine Aggregation Other Chemical Modifications Modified LDL Intima Modified LDL are Proinflammatory Steinberg D et al. N Engl J Med 1989;320: Slide Source: Lipids Online

57
HDL inhibit the oxidative modification of LDL
HDL inhibit the oxidative modification of LDL Vessel Lumen Monocyte LDL Adhesion Molecules HDL inhibit the oxidative modification of LDL HDL has protective effects in addition to promoting cholesterol efflux. One of the best known of these is the ability to inhibit the oxidation of LDL. To the extent that LDL oxidation is an important step in the development of the inflammatory process, this property of HDL is clearly anti-inflammatory. Inhibition of LDL oxidation by HDL: role of paraoxonase The importance of paraoxonase is now fairly well established. Paraoxonase is transported in plasma as a component of HDL, and it has also been shown in vitro in many studies that paraoxonase inhibits the oxidative modification of LDL. The working hypothesis of a number of groups is that the presence of paraoxonase in HDL may account for at least a proportion of the antioxidant properties of these lipoproteins, and thus contributes to HDL's anti-inflammatory effect. HDL has an antioxidant and anti-inflammatory effect beyond transporting paraoxonase. Cholesteryl ester transfer protein (CETP) transfers oxidized lipids from LDL to HDL. Once these oxidized lipids are in HDL, they are reduced by HDL apolipoproteins; this has been shown for both apo A-I and apo A-II. It has also been shown that the liver takes up reduced lipids from HDL more rapidly than from LDL. Therefore, HDL reduces the oxidized lipids in LDL, a further anti-inflammatory effect of HDL. Reference: Mackness MI, Abbott C, Arrol S, Durrington PN. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochem J 1993;294: Endothelium MCP-1 LDL HDL Inhibit Oxidation of LDL Modified LDL Cytokines Foam Cell Macrophage Intima HDL Promote Cholesterol Efflux Mackness MI et al. Biochem J 1993;294:
 transfers oxidized lipids from LDL to HDL. Once these oxidized lipids are in HDL, they are reduced by HDL apolipoproteins; this has been shown for both apo A-I and apo A-II. It has also been shown that the liver takes up reduced lipids from HDL more rapidly than from LDL. Therefore, HDL reduces the oxidized lipids in LDL, a further anti-inflammatory effect of HDL. Reference: Mackness MI, Abbott C, Arrol S, Durrington PN. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochem J 1993;294: Endothelium. MCP-1. LDL. HDL Inhibit Oxidation of LDL. Modified LDL. Cytokines. Foam Cell. Macrophage. Intima. HDL Promote Cholesterol Efflux. Mackness MI et al. Biochem J 1993;294:")
58
Reverse Cholesterol Transport
Reverse Cholesterol Transport HDL Metabolism and Reverse Cholesterol Transport Cholesterol that is synthesized or deposited in peripheral tissues is returned to the liver in a process referred to as reverse cholesterol transport in which high-density lipoprotein (HDL) plays a central role. HDL may be secreted by the liver or intestine in the form of nascent particles consisting of phospholipid and apolipoprotein A-I (apoA-I). Nascent HDL interacts with peripheral cells, such as macrophages, to facilitate the removal of excess free cholesterol (FC), a process facilitated by the ATP-binding cassette protein 1 (ABC1) gene. FC is generated in part by the hydrolysis of intracellular cholesteryl ester (CE) stores. HDL is then converted into mature CE–rich HDL as a result of the plasma cholesterol-esterifying enzyme lecithin:cholesterol acyltransferase (LCAT), which is activated by apoA-I. CE may be removed by several different pathways, including selective uptake by the liver, ie, the removal of lipid without the uptake of HDL proteins (shown in this slide). Selective uptake appears to be mediated by the scavenger receptor class-B, type I (SR-BI), which is expressed in the liver and has been shown to be a receptor for HDL. CE derived from HDL contributes to the hepatic–cholesterol pool used for bile acid synthesis. Cholesterol is eventually excreted from the body either as bile acid or as free cholesterol in the bile. References: Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228.
 plays a central role. HDL may be secreted by the liver or intestine in the form of nascent particles consisting of phospholipid and apolipoprotein A-I (apoA-I). Nascent HDL interacts with peripheral cells, such as macrophages, to facilitate the removal of excess free cholesterol (FC), a process facilitated by the ATP-binding cassette protein 1 (ABC1) gene. FC is generated in part by the hydrolysis of intracellular cholesteryl ester (CE) stores. HDL is then converted into mature CE–rich HDL as a result of the plasma cholesterol-esterifying enzyme lecithin:cholesterol acyltransferase (LCAT), which is activated by apoA-I. CE may be removed by several different pathways, including selective uptake by the liver, ie, the removal of lipid without the uptake of HDL proteins (shown in this slide). Selective uptake appears to be mediated by the scavenger receptor class-B, type I (SR-BI), which is expressed in the liver and has been shown to be a receptor for HDL. CE derived from HDL contributes to the hepatic–cholesterol pool used for bile acid synthesis. Cholesterol is eventually excreted from the body either as bile acid or as free cholesterol in the bile. References: Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228.")
59
Statins: Mechanism of Action
Statins: Mechanism of Action VLDL Cholesterol synthesis VLDLR LDL receptor (B–E receptor) synthesis Apo B LDL receptor–mediated hepatic uptake of LDL and VLDL remnants Apo E Statins: mechanism of action As inhibitors of hepatic HMG-CoA reductase, the enzyme catalyzing the rate-limiting step in hepatic cholesterol synthesis, statins decrease synthesis of cholesterol by the liver, which results in two important effects: the up-regulation of LDL receptors by hepatocytes and consequent increased removal of apolipoprotein (apo) E– and B–containing lipoproteins from the circulation, and a reduction in the synthesis and secretion of lipoproteins from the liver. The net effect of statin therapy is to lower plasma concentrations of cholesterol-carrying lipoproteins, the most prominent of which is LDL. Importantly, however, statins also increase the removal and reduce the secretion of remnant particles, i.e., very low density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL). This means that in patients who have an elevation of both LDL-C and triglycerides (indicating increased levels of triglyceride-rich VLDL and IDL remnants as well as LDL), a statin is one of the therapies of choice because of its ability to effectively lower LDL-C and non–high-density lipoprotein cholesterol (non-HDL-C) levels. Intracellular Cholesterol Serum LDL-C LDL Apo B Serum VLDL remnants Serum IDL Hepatocyte Systemic Circulation Reduce hepatic cholesterol synthesis, lowering intracellular cholesterol, which stimulates upregulation of LDL receptor and increases the uptake of non-HDL particles from the systemic circulation.
 synthesis. Apo B. LDL receptor–mediated hepatic uptake of LDL and VLDL remnants. Apo E. Statins: mechanism of action. As inhibitors of hepatic HMG-CoA reductase, the enzyme catalyzing the rate-limiting step in hepatic cholesterol synthesis, statins decrease synthesis of cholesterol by the liver, which results in two important effects: the up-regulation of LDL receptors by hepatocytes and consequent increased removal of apolipoprotein (apo) E– and B–containing lipoproteins from the circulation, and a reduction in the synthesis and secretion of lipoproteins from the liver. The net effect of statin therapy is to lower plasma concentrations of cholesterol-carrying lipoproteins, the most prominent of which is LDL. Importantly, however, statins also increase the removal and reduce the secretion of remnant particles, i.e., very low density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL). This means that in patients who have an elevation of both LDL-C and triglycerides (indicating increased levels of triglyceride-rich VLDL and IDL remnants as well as LDL), a statin is one of the therapies of choice because of its ability to effectively lower LDL-C and non–high-density lipoprotein cholesterol (non-HDL-C) levels. Intracellular Cholesterol. Serum LDL-C. LDL. Apo B. Serum VLDL remnants. Serum IDL. Hepatocyte. Systemic Circulation. Reduce hepatic cholesterol synthesis, lowering intracellular cholesterol, which stimulates upregulation of LDL receptor and increases the uptake of non-HDL particles from the systemic circulation.")
60
Nicotinic Acid: Mechanism of Action
Nicotinic Acid: Mechanism of Action Mobilization of FFA Apo B Serum VLDL results in reduced lipolysis to LDL VLDL VLDL TG synthesis VLDL secretion Serum LDL Nicotinic acid: mechanism of action The last of our LDL-C–lowering drugs is nicotinic acid, or niacin. Niacin appears to exert its effects by inhibiting lipoprotein synthesis and decreasing the production of VLDL particles by the liver. It inhibits the peripheral mobilization of free fatty acids, thus reducing hepatic synthesis of triglycerides and the secretion of VLDL. It also reduces apo B. The net result is a reduction in VLDL particles secreted by the liver and thus less substrate to make LDL particles. It increases the production of apo A-I and thereby HDL through mechanisms that are not clear. LDL HDL Liver Circulation Hepatocyte Systemic Circulation Decreases hepatic production of VLDL and of apo B

61
Severe hypertriglyceridemia and chylomicronemia (type V hyperlipidemia) - eruptive xanthomata A 36 year old moderate drinker with diabetic ketoacidosis and acute pancreatitis. Eruptive xanthomata in severe hypertriglyceridemia and chylomicronemia (type V hyperlipidemia) due to uncontrolled diabetes mellitus (diabetic lipemia) and to alcohol abuse.
 due to uncontrolled diabetes mellitus (diabetic lipemia) and to alcohol abuse.")
65
High-density lipoprotein subspecies
High-density lipoprotein subspecies Figure High-density lipoprotein (HDL) subspecies. HDL can be separated into several subclasses on the basis of size, density, and apolipoprotein (apo) composition. A, HDL has been separated schematically into HDL-3 (smaller and more dense) and HDL-2 (larger and less dense) by size or density alone. Many studies have suggested that HDL-2 is the HDL fraction that is most protective against the development of coronary heart disease. Another way of separating HDL subspecies is to divide HDL into HDL particles that contain apo A-I without apo A-II (Lp A-I) and those that contain both apo A-I and apo A-II (Lp A-I/A-II). Lp A-I contains at least two distinct types of particles, one in the HDL-3 subclass and another in the HDL-2 subclass. Lp A-I/A-II contains at least three distinct types of particles: one lies in the HDL-3 subclass, another in the HDL-2 subclass, and the third lies intermediate between the two. Using this classification, Lp A-I has been suggested to be antiatherogenic and Lp A-I/A-II either neutral or actually atherogenic. B, Bottom densities of 1.30 g/mL with an overlay with physiologic saline provide optimal separation of HDL from the other lipoproteins using a 70-Ti angled head rotor and a single 4-h spin. The HDL profile shown here demonstrates two HDL subspecies, HDL-3 and HDL-2. C (inset), HDL-vertical auto profile density gradient ultracentrifugation separation of HDL subspecies uses a bottom density of 1.21 g/mL and an overlay of 1.06 g/mL and a VTi80 vertical rotor centrifuged at 80,000 rpm for 90 minutes; the analysis of cholesterol is by a continuous flow method []. The resulting profile is superior to that obtained with a 70-Ti angled head rotor; three to five peaks can be identified. HDL single vertical spin preparative fractionation of HDL sub-species from the same plasma sample used in the inset is shown in C. The resolution is comparable with, or superior to, that shown in the inset. (Part C adapted from Cheung et al. [].)
 subspecies. HDL can be separated into several subclasses on the basis of size, density, and apolipoprotein (apo) composition. A, HDL has been separated schematically into HDL-3 (smaller and more dense) and HDL-2 (larger and less dense) by size or density alone. Many studies have suggested that HDL-2 is the HDL fraction that is most protective against the development of coronary heart disease. Another way of separating HDL subspecies is to divide HDL into HDL particles that contain apo A-I without apo A-II (Lp A-I) and those that contain both apo A-I and apo A-II (Lp A-I/A-II). Lp A-I contains at least two distinct types of particles, one in the HDL-3 subclass and another in the HDL-2 subclass. Lp A-I/A-II contains at least three distinct types of particles: one lies in the HDL-3 subclass, another in the HDL-2 subclass, and the third lies intermediate between the two. Using this classification, Lp A-I has been suggested to be antiatherogenic and Lp A-I/A-II either neutral or actually atherogenic. B, Bottom densities of 1.30 g/mL with an overlay with physiologic saline provide optimal separation of HDL from the other lipoproteins using a 70-Ti angled head rotor and a single 4-h spin. The HDL profile shown here demonstrates two HDL subspecies, HDL-3 and HDL-2. C (inset), HDL-vertical auto profile density gradient ultracentrifugation separation of HDL subspecies uses a bottom density of 1.21 g/mL and an overlay of 1.06 g/mL and a VTi80 vertical rotor centrifuged at 80,000 rpm for 90 minutes; the analysis of cholesterol is by a continuous flow method []. The resulting profile is superior to that obtained with a 70-Ti angled head rotor; three to five peaks can be identified. HDL single vertical spin preparative fractionation of HDL sub-species from the same plasma sample used in the inset is shown in C. The resolution is comparable with, or superior to, that shown in the inset. (Part C adapted from Cheung et al. [].)")
66
Lipoprotein (a) Figure Lipoprotein (a) or [Lp(a)]. This variant lipoprotein particle is present in varying levels in different ethnic populations. A, Lp(a) consists of low-density lipoprotein particles with an extra protein called apo (a) apparently covalently associated with apo B-100. Apo(a) contains repeated structural regions, called kringles, homologous to those contained in the fibrinolytic enzyme plasminogen. In several studies, Lp(a) has been associated with a greater risk of coronary heart disease, and also with accelerated restenosis of coronary artery bypass grafts. Conflicting data, however, also exist. The relationship to atherosclerosis is complex and only partly related to the levels of Lp(a), which appear to be largely genetically determined. Diet and most medications have little effect on circulating levels of Lp(a). The mechanism for the atherogenicity of Lp(a) may be related to its homology to plasminogen and involve an aberration in the thrombotic system. B, Images of Lp(a) obtained by scanning tunneling electron microscopy. (Part B from Xu []; with permission.)
 or [Lp(a)]. This variant lipoprotein particle is present in varying levels in different ethnic populations. A, Lp(a) consists of low-density lipoprotein particles with an extra protein called apo (a) apparently covalently associated with apo B-100. Apo(a) contains repeated structural regions, called kringles, homologous to those contained in the fibrinolytic enzyme plasminogen. In several studies, Lp(a) has been associated with a greater risk of coronary heart disease, and also with accelerated restenosis of coronary artery bypass grafts. Conflicting data, however, also exist. The relationship to atherosclerosis is complex and only partly related to the levels of Lp(a), which appear to be largely genetically determined. Diet and most medications have little effect on circulating levels of Lp(a). The mechanism for the atherogenicity of Lp(a) may be related to its homology to plasminogen and involve an aberration in the thrombotic system. B, Images of Lp(a) obtained by scanning tunneling electron microscopy. (Part B from Xu []; with permission.)")
68
Regulation of cholesterol biosynthesis
Regulation of cholesterol biosynthesis Figure Regulation of cholesterol biosynthesis. At a cellular level, cholesterol biosynthesis is regulated by intracellular free cholesterol. When this pool is reduced, the number of receptors bringing cholesterol into the cell is increased and cholesterol biosynthesis is increased. When the intracellular cholesterol is increased, the number of receptors is decreased, as is cholesterol biosynthesis. Dietary fiber, by reducing cholesterol absorption in the gastrointestinal tract and removing it from the body, reduces intracellular cholesterol and triggers an increase in cholesterol biosynthesis. Therefore, the observed reduction in total cholesterol due to dietary fiber is increased when it is given in combination with an inhibitor of cholesterol biosynthesis. LDL—low-density lipoprotein.

69
Apolipoprotein B-100 on low-density lipoprotein
Apolipoprotein B-100 on low-density lipoprotein Figure 6-2. Representation of apolipoprotein (apo) B-100 on low-density lipoprotein (LDL). Apo B-100 consists of 4536 amino acids (molecular weight, approximately 540,000 D) and weaves in and out of the LDL surface. Apo B is susceptible to proteolysis (especially by thrombin and kallikrein) during isolation. The thrombin cleavage sites are represented in the figure at amino acid 1297 and The amphipathic nature of apo B makes some regions buried in the core and some exposed to the outside. Isolated apo B is very insoluble in aqueous solutions. There are at least 20 potential N-glycosylation sites and several heparin binding sites. The receptor-binding domain of apo B appears to be located near amino acid Apo B does not exchange among lipoproteins. (Adapted from Yang et al. [].)
 B-100 on low-density lipoprotein (LDL). Apo B-100 consists of 4536 amino acids (molecular weight, approximately 540,000 D) and weaves in and out of the LDL surface. Apo B is susceptible to proteolysis (especially by thrombin and kallikrein) during isolation. The thrombin cleavage sites are represented in the figure at amino acid 1297 and The amphipathic nature of apo B makes some regions buried in the core and some exposed to the outside. Isolated apo B is very insoluble in aqueous solutions. There are at least 20 potential N-glycosylation sites and several heparin binding sites. The receptor-binding domain of apo B appears to be located near amino acid Apo B does not exchange among lipoproteins. (Adapted from Yang et al. [].)")
70
Lipoprotein(a) Figure Lipoprotein(a) [Lp(a)] represents a distinct genetic form of low-density lipoprotein (LDL) wherein apolipoprotein (a) [apo(a)] is linked to apo B-100 via cysteine K436 of apo(a) and an unknown cysteine from the carboxyl end of apo B by disulfide linkage. The apo(a) is a glycoprotein that exhibits considerable genetically determined polymorphism among individuals producing a wide range of molecular weights, from 300 to 800 kD. Apo(a) is strikingly similar to plasminogen. Although three of the five kringles present in plasminogen (K1, K2, and K3) are absent in apo(a), K4 is repeated several times. A number of epidemiologic studies have shown a link between elevated levels of Lp(a) and cardiovascular disease []. It may have both atherogenic and antithrombolytic potential. Apo(a) has been found in the macrophage-rich atherosclerotic lesion. The mechanism(s) by which Lp(a) might contribute to cardiovascular diseases is currently theoretical. Recently, mice strains that overexpress human apo(a) have been developed []. These animals develop accelerated atherosclerosis when fed atherogenic diets. (Adapted from Scanu and Fless [].)
 [Lp(a)] represents a distinct genetic form of low-density lipoprotein (LDL) wherein apolipoprotein (a) [apo(a)] is linked to apo B-100 via cysteine K436 of apo(a) and an unknown cysteine from the carboxyl end of apo B by disulfide linkage. The apo(a) is a glycoprotein that exhibits considerable genetically determined polymorphism among individuals producing a wide range of molecular weights, from 300 to 800 kD. Apo(a) is strikingly similar to plasminogen. Although three of the five kringles present in plasminogen (K1, K2, and K3) are absent in apo(a), K4 is repeated several times. A number of epidemiologic studies have shown a link between elevated levels of Lp(a) and cardiovascular disease []. It may have both atherogenic and antithrombolytic potential. Apo(a) has been found in the macrophage-rich atherosclerotic lesion. The mechanism(s) by which Lp(a) might contribute to cardiovascular diseases is currently theoretical. Recently, mice strains that overexpress human apo(a) have been developed []. These animals develop accelerated atherosclerosis when fed atherogenic diets. (Adapted from Scanu and Fless [].)")
71
Foam cell formation Figure Foam cell formation. Foam cells derive their name by the foamy appearance caused by lipid accumulation and are the hallmark of early atherosclerotic, fatty streak lesions. The predominant cell type that accumulates lipids is the macrophage, although smooth muscle cell-derived foam cells also occur. Increased chemotactic activity may account for the presence of monocytes or macrophages in the artery. However, the factors that aid in the differentiation of monocytes into differentiated tissue macrophages are yet unknown. Modified lipoproteins are recognized and internalized by the scavenger receptors on the macrophages; there is no feedback regulation of the uptake by this mechanism. The accumulated cellular cholesterol is readily converted to cholesteryl ester and stored as large cytoplasmic lipid droplets. Recently, methods have been developed to isolate and study foam cells from the atherosclerotic artery []. (Adapted from Grundy [].)
")
72
Types of oxidized low-density lipoprotein
Types of oxidized low-density lipoprotein Figure Types of oxidized low-density lipoprotein (LDL) (A and B). (Adapted from Parthasarathy et al. [].)
 (A and B). (Adapted from Parthasarathy et al. [].)")
73
Low-density lipoprotein may enter the artery wall and be trapped by interactions with extracellular matrix components Figure A, Elevated levels of plasma low-density lipoprotein (LDL) alone may be sufficient to initiate the fatty streak lesion. High levels of plasma LDL may increase the availability of LDL in the intima. Free or matrix-bound LDL may undergo oxidation by cells such as endothelial cells (EC), smooth muscle cells (SMCs), or macrophages (MOs). The mildly oxidized LDL may increase the chemotactic recruitment of monocytes by inducing the expression of monocyte chemotactic protein-1 (MCP-1) or by generating lysophospholipids (more extensively, oxidized LDL). Monocytes may differentiate into macrophages, and components of oxidized LDL may promote retention of macrophages in the artery. Macrophages and oxidized LDL may then interact via scavenger pathways leading to foam cell formation. B, LDL may enter the artery wall and may be trapped by specific interactions with extracellular matrix components. Cells of the artery may inhibit the oxidation of LDL by mechanisms not yet characterized. Superoxide generation, 15-lipoxygenase activity, and other pathways may contribute to the oxidation. Minimally oxidized LDL (with intact apolipoprotein (apo) B-100) possesses components that have potent biologic properties; these include their ability to induce MCP-1 and granulocyte macrophage colony-stimulating factor. Further increase in oxidation generates oxidized LDL in which apo B-100 is extremely proteolyzed and altered. The maximally oxidized LDL also possesses components that are chemotactic to monocytes and T lymphocytes. Lipids from oxidized LDL also inhibit the chemotaxis of differential macrophages. Macrophages take up oxidized LDL via scavenger receptors and accumulate cholesterol esters, thus accounting for foam cells. There are over 20 postulated mechanisms by which the oxidized LDL may contribute to atherogenesis. Most of the proatherogenic effects of oxidized LDL are mediated by its lipid components. (Adapted from Steinberg [] and Quinn et al. [].)
 alone may be sufficient to initiate the fatty streak lesion. High levels of plasma LDL may increase the availability of LDL in the intima. Free or matrix-bound LDL may undergo oxidation by cells such as endothelial cells (EC), smooth muscle cells (SMCs), or macrophages (MOs). The mildly oxidized LDL may increase the chemotactic recruitment of monocytes by inducing the expression of monocyte chemotactic protein-1 (MCP-1) or by generating lysophospholipids (more extensively, oxidized LDL). Monocytes may differentiate into macrophages, and components of oxidized LDL may promote retention of macrophages in the artery. Macrophages and oxidized LDL may then interact via scavenger pathways leading to foam cell formation. B, LDL may enter the artery wall and may be trapped by specific interactions with extracellular matrix components. Cells of the artery may inhibit the oxidation of LDL by mechanisms not yet characterized. Superoxide generation, 15-lipoxygenase activity, and other pathways may contribute to the oxidation. Minimally oxidized LDL (with intact apolipoprotein (apo) B-100) possesses components that have potent biologic properties; these include their ability to induce MCP-1 and granulocyte macrophage colony-stimulating factor. Further increase in oxidation generates oxidized LDL in which apo B-100 is extremely proteolyzed and altered. The maximally oxidized LDL also possesses components that are chemotactic to monocytes and T lymphocytes. Lipids from oxidized LDL also inhibit the chemotaxis of differential macrophages. Macrophages take up oxidized LDL via scavenger receptors and accumulate cholesterol esters, thus accounting for foam cells. There are over 20 postulated mechanisms by which the oxidized LDL may contribute to atherogenesis. Most of the proatherogenic effects of oxidized LDL are mediated by its lipid components. (Adapted from Steinberg [] and Quinn et al. [].)")
74
Scavenger receptor (types I and II)
Scavenger receptor (types I and II) Figure Scavenger receptor (type I and type II). The scavenger receptor is biochemically and genetically distinct from the low-density lipoprotein (LDL) receptor. It is expressed in macrophages, endothelial cells, and smooth muscle cells. It recognizes acetyl LDL, oxidized LDL, and several polyanionic molecules such as polyinosinic acid, fucoidan, and bacterial lipopolysaccharide. Unlike the LDL receptor, the expression of the scavenger receptor is not regulated by cellular cholesterol, thus permitting massive uptake of cholesterol when modified lipoproteins are presented to the cell. The presence of this receptor has been demonstrated in human and rabbit atherosclerotic lesions. The type I scavenger receptor has six domains and 453 amino acids. The protein is made of domain I, a 50-amino-acid N-terminal cytoplasmic domain; domain II, a 26-amino-acid transmembrane domain; and extracellular domains III, IV, V, and VI consisting of 32, 163, 72, and 110 amino acids, respectively. Domain IV has a distinguishing feature of heptad repeat units. Domain V has collagen-like repeats of Gly-X-Y sequences and is presumably involved in ligand binding. Domain VI is a cysteine-rich domain that is absent in the type II receptor. The receptor in the reduced form is a 77-kD glycoprotein that in the unreduced form forms a 220-kD ligand-binding trimer. It is suggested that additional scavenger receptors that bind to oxidized LDL are present in macrophages. TM - thrombomodulin. (Adapted from Kodama et al. [] and Rohrer et al. [].)
 Figure Scavenger receptor (type I and type II). The scavenger receptor is biochemically and genetically distinct from the low-density lipoprotein (LDL) receptor. It is expressed in macrophages, endothelial cells, and smooth muscle cells. It recognizes acetyl LDL, oxidized LDL, and several polyanionic molecules such as polyinosinic acid, fucoidan, and bacterial lipopolysaccharide. Unlike the LDL receptor, the expression of the scavenger receptor is not regulated by cellular cholesterol, thus permitting massive uptake of cholesterol when modified lipoproteins are presented to the cell. The presence of this receptor has been demonstrated in human and rabbit atherosclerotic lesions. The type I scavenger receptor has six domains and 453 amino acids. The protein is made of domain I, a 50-amino-acid N-terminal cytoplasmic domain; domain II, a 26-amino-acid transmembrane domain; and extracellular domains III, IV, V, and VI consisting of 32, 163, 72, and 110 amino acids, respectively. Domain IV has a distinguishing feature of heptad repeat units. Domain V has collagen-like repeats of Gly-X-Y sequences and is presumably involved in ligand binding. Domain VI is a cysteine-rich domain that is absent in the type II receptor. The receptor in the reduced form is a 77-kD glycoprotein that in the unreduced form forms a 220-kD ligand-binding trimer. It is suggested that additional scavenger receptors that bind to oxidized LDL are present in macrophages. TM - thrombomodulin. (Adapted from Kodama et al. [] and Rohrer et al. [].)")
75
Low-density lipoprotein and scavenger receptor activities as a function of time in culture Figure Low-density lipoprotein (LDL) and scavenger receptor activities as a function of time in culture. A, LDL and minimally oxidized LDL are recognized by the LDL receptor. Incubation of macrophages with these lipoproteins does not result in lipid accumulation. Further oxidation results in poor recognition by the LDL receptor. More extensively oxidized LDL is recognized by the macrophage scavenger receptors and other uncharacterized receptors. Extensively oxidized LDL may also be recognized by CD36, FcγR11B2, and several other surface proteins. During the differentiation of monocytes in culture, scavenger receptors are induced. The relative contribution of different scavenger receptors in the uptake of oxidized LDL has not been established. B, Freshly isolated monocytes do not have a high degree of expression of the acetyl LDL receptor, and they take up modified lipoproteins poorly. Upon culturing, they take up increased amounts of the acetyl LDL, and the cellular expression of the receptor increases. Cultured THP-1 line of monocytes can be induced to express scavenger receptor (acetyl LDL receptor) by exposure to differentiating agents such as phorbol myristate acetate (PMA). Smooth muscle cells and fibroblasts can also be induced to express the acetyl LDL receptor upon suitable stimulation. apo - apolipoprotein; MDA - malondialdehyde. (Part A adapted from Fogelman et al. [].)
 and scavenger receptor activities as a function of time in culture. A, LDL and minimally oxidized LDL are recognized by the LDL receptor. Incubation of macrophages with these lipoproteins does not result in lipid accumulation. Further oxidation results in poor recognition by the LDL receptor. More extensively oxidized LDL is recognized by the macrophage scavenger receptors and other uncharacterized receptors. Extensively oxidized LDL may also be recognized by CD36, FcγR11B2, and several other surface proteins. During the differentiation of monocytes in culture, scavenger receptors are induced. The relative contribution of different scavenger receptors in the uptake of oxidized LDL has not been established. B, Freshly isolated monocytes do not have a high degree of expression of the acetyl LDL receptor, and they take up modified lipoproteins poorly. Upon culturing, they take up increased amounts of the acetyl LDL, and the cellular expression of the receptor increases. Cultured THP-1 line of monocytes can be induced to express scavenger receptor (acetyl LDL receptor) by exposure to differentiating agents such as phorbol myristate acetate (PMA). Smooth muscle cells and fibroblasts can also be induced to express the acetyl LDL receptor upon suitable stimulation. apo - apolipoprotein; MDA - malondialdehyde. (Part A adapted from Fogelman et al. [].)")
76
The atherosclerotic process
The atherosclerotic process Figure The atherosclerotic process. A, Artery depicting early fatty streak development. B, 1, low-density lipoproteins (LDL) becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)
 becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)")
77
The atherosclerotic process
The atherosclerotic process Figure The atherosclerotic process. A, Artery depicting early fatty streak development. B, 1, low-density lipoproteins (LDL) becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)
 becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)")
78
The atherosclerotic process
The atherosclerotic process Figure The atherosclerotic process. A, Artery depicting early fatty streak development. B, 1, low-density lipoproteins (LDL) becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)
 becomes oxidized within the arterial subendothelial space. 2, Circulating monocytes are recruited to the subendothelial space by chemoattractants, including oxidized LDL. 3, These monocytes undergo differentiation, becoming macrophages, which are scavenger cells that recognize and accumulate oxidized LDL. 4, The lipid-laden macrophages then become foam cells, which cluster under the endothelial lining to form a bulge into the artery. 5, This bulge is called a fatty streak and is the first overt sign of atherosclerotic change. C, Cross-section of an artery with an atherosclerotic lesion with a narrowed lumen. (Courtesy of Merrell Dow Pharmaceuticals Inc., Cincinnati, OH.)")
79
Overview of high-density lipoprotein metabolism and reverse cholesterol transport Figure 7-9. Overview of high-density lipoprotein (HDL) metabolism and reverse cholesterol transport. Four major pathways are involved in the synthesis of mature HDL. Nascent or pre-HDL, which are composed primarily of apolipoprotein (apo) A-I phospholipid disks, are secreted from the human intestine and liver. Lipids and apolipoprotein constituents of HDL are acquired from the intravascular metabolism and remodeling of both-triglyceride rich chylomicrons and hepatic very low-density lipoprotein (VLDL), which converts nascent HDL to mature HDL. Nascent HDL plays a pivotal role in lipoprotein metabolism and reverse cholesterol transport by facilitating the efflux of excess cholesterol from the membranes of peripheral cells including macrophages by interaction with the ABCA1 transporter []. The free cholesterol on nascent HDL is esterified to cholesteryl esters by lecithin cholesterol acyltransferase (LCAT). With the formation of cholesteryl esters, the nascent HDL are converted to spherical lipoproteins with a hydrated density of HDL3. HDL3 are converted to the larger HDL2 by the acquisition of lipids and apolipoproteins (eg, apo C-III) released during the stepwise delipidation and remodeling of the triglyceride-rich chylomicrons and VLDL and by the esterification of the cholesterol removed from peripheral tissues. HDL transports cholesterol back to the liver by two pathways. The first pathway involves a direct delivery of cholesterol to the liver by a newly recognized receptor, SR-BI [][][], that functions to remove cholesteryl esters selectively from lipoproteins without holoparticle uptake and degradation. Additionally, HDL particles are taken up intact and degraded by receptors primarily in the liver and kidney. In the second pathway, HDL cholesteryl esters are exchanged for triglycerides in the apo B-containing lipoproteins (VLDL, intermediate-density lipoprotein [IDL], low-density lipoprotein [LDL]) by the cholesteryl ester transfer protein (CETP) []. A significant fraction of cholesteryl esters present in HDL are transferred back to the liver by the LDL pathway. Thus, cholesterol may be transported back to the liver directly by HDL or following exchange to VLDL-IDL-LDL. It also has been proposed that a variable portion of tissue cholesterol is transported to the liver by HDL particles containing apo E, which may interact with both the hepatic LRP(LDL receptor-related protein) and LDL receptors (LDL-R). HDL-R--high-density lipoprotein receptors; HL--hepatic lipase; LPL--lipoprotein lipase.
 metabolism and reverse cholesterol transport. Four major pathways are involved in the synthesis of mature HDL. Nascent or pre-HDL, which are composed primarily of apolipoprotein (apo) A-I phospholipid disks, are secreted from the human intestine and liver. Lipids and apolipoprotein constituents of HDL are acquired from the intravascular metabolism and remodeling of both-triglyceride rich chylomicrons and hepatic very low-density lipoprotein (VLDL), which converts nascent HDL to mature HDL. Nascent HDL plays a pivotal role in lipoprotein metabolism and reverse cholesterol transport by facilitating the efflux of excess cholesterol from the membranes of peripheral cells including macrophages by interaction with the ABCA1 transporter []. The free cholesterol on nascent HDL is esterified to cholesteryl esters by lecithin cholesterol acyltransferase (LCAT). With the formation of cholesteryl esters, the nascent HDL are converted to spherical lipoproteins with a hydrated density of HDL3. HDL3 are converted to the larger HDL2 by the acquisition of lipids and apolipoproteins (eg, apo C-III) released during the stepwise delipidation and remodeling of the triglyceride-rich chylomicrons and VLDL and by the esterification of the cholesterol removed from peripheral tissues. HDL transports cholesterol back to the liver by two pathways. The first pathway involves a direct delivery of cholesterol to the liver by a newly recognized receptor, SR-BI [][][], that functions to remove cholesteryl esters selectively from lipoproteins without holoparticle uptake and degradation. Additionally, HDL particles are taken up intact and degraded by receptors primarily in the liver and kidney. In the second pathway, HDL cholesteryl esters are exchanged for triglycerides in the apo B-containing lipoproteins (VLDL, intermediate-density lipoprotein [IDL], low-density lipoprotein [LDL]) by the cholesteryl ester transfer protein (CETP) []. A significant fraction of cholesteryl esters present in HDL are transferred back to the liver by the LDL pathway. Thus, cholesterol may be transported back to the liver directly by HDL or following exchange to VLDL-IDL-LDL. It also has been proposed that a variable portion of tissue cholesterol is transported to the liver by HDL particles containing apo E, which may interact with both the hepatic LRP(LDL receptor-related protein) and LDL receptors (LDL-R). HDL-R--high-density lipoprotein receptors; HL--hepatic lipase; LPL--lipoprotein lipase.")
80
ABCA1 transporter Figure The ABCA1 transporter. Poorly lipidated apolipoprotein (apo) A1 interacts with the ABCA1 transporter to facilitate the removal of excess cholesterol from peripheral cells. The nascent high-density lipoprotein (HDL) formed following the interaction of apo A1 with the ABCA1 transporter on the cell membrane is converted to mature HDL by the LCAT (A). The genetic defect in Tangier disease (B) is a structural mutation in the ABCA1 transporter. The defect in the ABCA1 transporter results in decreased efflux of cholesterol from the cell and reduced lipidation of apo A1. The poorly lipidated HDL is rapidly degraded by the kidney, leading to the low plasma HDL levels characteristic of Tangier disease [].
 A1 interacts with the ABCA1 transporter to facilitate the removal of excess cholesterol from peripheral cells. The nascent high-density lipoprotein (HDL) formed following the interaction of apo A1 with the ABCA1 transporter on the cell membrane is converted to mature HDL by the LCAT (A). The genetic defect in Tangier disease (B) is a structural mutation in the ABCA1 transporter. The defect in the ABCA1 transporter results in decreased efflux of cholesterol from the cell and reduced lipidation of apo A1. The poorly lipidated HDL is rapidly degraded by the kidney, leading to the low plasma HDL levels characteristic of Tangier disease [].")
81
ABCA1 transporter Figure The ABCA1 transporter. Poorly lipidated apolipoprotein (apo) A1 interacts with the ABCA1 transporter to facilitate the removal of excess cholesterol from peripheral cells. The nascent high-density lipoprotein (HDL) formed following the interaction of apo A1 with the ABCA1 transporter on the cell membrane is converted to mature HDL by the LCAT (A). The genetic defect in Tangier disease (B) is a structural mutation in the ABCA1 transporter. The defect in the ABCA1 transporter results in decreased efflux of cholesterol from the cell and reduced lipidation of apo A1. The poorly lipidated HDL is rapidly degraded by the kidney, leading to the low plasma HDL levels characteristic of Tangier disease [].
 A1 interacts with the ABCA1 transporter to facilitate the removal of excess cholesterol from peripheral cells. The nascent high-density lipoprotein (HDL) formed following the interaction of apo A1 with the ABCA1 transporter on the cell membrane is converted to mature HDL by the LCAT (A). The genetic defect in Tangier disease (B) is a structural mutation in the ABCA1 transporter. The defect in the ABCA1 transporter results in decreased efflux of cholesterol from the cell and reduced lipidation of apo A1. The poorly lipidated HDL is rapidly degraded by the kidney, leading to the low plasma HDL levels characteristic of Tangier disease [].")
82
Plasma lipid exchange Figure Plasma lipid exchange. There is a dynamic exchange of plasma lipids that occurs continuously in the plasma. Cholesterol ester transfer protein (CETP) mediates this process. In the setting of increased amounts of triglyceride-rich lipoproteins, either very low-density lipoprotein (VLDL) or chylomicron, triglyceride (TG) is exchanged for cholesterol esters (CE). Both of these lipids are hydrophobic and reside in the core of the lipoproteins. Triglyceride, but not cholesterol ester, is a substrate for the actions of lipoprotein lipase and hepatic lipase, the two most important triglyceride-degrading enzymes in the plasma. Therefore, lipoproteins that become triglyceride enriched can be converted to smaller particles because this core lipid is metabolism to fatty acids. This is the mechanism responsible for the production of smaller dense LDL and HDL3 in hypertriglyceridemia subjects, like those with diabetes. Moreover, because of this metabolic association triglyceride, LDL size and HDL cholesterol are metabolically linked. Thus, individual contributions of each of these factors to cardiovascular risk are confounded by coordinated changes in the other two lipoprotein parameters.
 mediates this process. In the setting of increased amounts of triglyceride-rich lipoproteins, either very low-density lipoprotein (VLDL) or chylomicron, triglyceride (TG) is exchanged for cholesterol esters (CE). Both of these lipids are hydrophobic and reside in the core of the lipoproteins. Triglyceride, but not cholesterol ester, is a substrate for the actions of lipoprotein lipase and hepatic lipase, the two most important triglyceride-degrading enzymes in the plasma. Therefore, lipoproteins that become triglyceride enriched can be converted to smaller particles because this core lipid is metabolism to fatty acids. This is the mechanism responsible for the production of smaller dense LDL and HDL3 in hypertriglyceridemia subjects, like those with diabetes. Moreover, because of this metabolic association triglyceride, LDL size and HDL cholesterol are metabolically linked. Thus, individual contributions of each of these factors to cardiovascular risk are confounded by coordinated changes in the other two lipoprotein parameters.")
83
A model depicting how lipoproteins may contribute to atherosclerosis
A model depicting how lipoproteins may contribute to atherosclerosis A model depicting how lipoproteins may contribute to atherosclerosis. At high concentrations of low density lipoproteins (LDL) or other atherogenic lipoproteins, the particles accumulate in the subendothelial space. The trapped LDL particles become oxidized, probably as a result of interactions with reactive oxygen species (ROS) produced by vascular cells. In vitro and in vivo studies suggest that such minimally modified LDL (MM-LDL) exhibits a potent biologic activity capable of inducing endothelial cells to express adhesion molecules for monocytes (X-LAM), monocyte chemotactic protein-1 (MCP-1), and macrophage-colony stimulating factor (M-CSF). The expression of these and other molecules results in the recruitment of blood monocytes to the artery wall, where the monocytes differentiate into macrophages. With time, the LDL particles become highly oxidized (Ox-LDL), perhaps as a result of the high levels of ROS produced by macrophages. Such highly oxidized LDL particles are recognized by scavenger receptors on macrophages, resulting in rapid endocytosis. Unlike the LDL receptor, the scavenger receptors are not down-regulated by high levels of cellular cholesterol, and the macrophages continue to accumulate cholesterol until they give rise to cholesterol-engorged foam cells. Such foam cells, the hallmark of fatty streaks, may contribute to the development of advanced lesions by the production of cytokines and growth factors such as M-CSF and interleukin-1 (IL-1). Many aspects of this model have been validated in vivo using animal models. (Adapted from Berliner and Haberland [9], with permission.)
 or. other atherogenic lipoproteins, the particles accumulate in the subendothelial space. The trapped LDL particles become oxidized, probably as a result of interactions with reactive oxygen species (ROS) produced by vascular cells. In vitro and in vivo studies suggest that such minimally. modified LDL (MM-LDL) exhibits a potent biologic activity capable of inducing endothelial cells to express adhesion molecules for monocytes (X-LAM), monocyte chemotactic protein-1 (MCP-1), and macrophage-colony stimulating factor (M-CSF). The expression of these and other molecules results in the recruitment of blood monocytes to the artery wall, where the monocytes differentiate into macrophages. With time, the LDL particles become highly oxidized (Ox-LDL), perhaps as a result of the high levels of ROS produced by macrophages. Such highly oxidized LDL particles are recognized by scavenger receptors on macrophages, resulting in rapid endocytosis. Unlike the LDL receptor, the scavenger receptors are not down-regulated by high levels of cellular cholesterol, and the macrophages continue to accumulate cholesterol until they give rise to cholesterol-engorged foam cells. Such foam cells, the hallmark of fatty streaks, may contribute to the development of advanced lesions by the production of cytokines and growth factors such as M-CSF and interleukin-1 (IL-1). Many aspects of this model. have been validated in vivo using animal models. (Adapted from Berliner and Haberland [9], with permission.)")
84
Liporprotein içindeki lipidlerin kimyasal yapısı
Liporprotein içindeki lipidlerin kimyasal yapısı Figure 4-6. General chemical structures of the lipids present in lipoprotein species. The lipids present in essentially all lipoprotein species are cholesterol, cholesteryl ester, phospholipids, and triglycerides. A, R1, R2, and R3 represent fatty acyl chains attached to the glycerol backbone to form triglyceride. B, Structure of cholesterol. Cholesteryl esters can be formed by attachment of a fatty acid to the hydroxyl group on carbon 3. C, The general structure of phospholipids, in which various head groups can combine with the phosphatidate; variation in the polar head groups of phospholipids results in neutral, acidic, or basic functional groups. (Adapted from Schreiber [].)
")
Benzer bir sunumlar
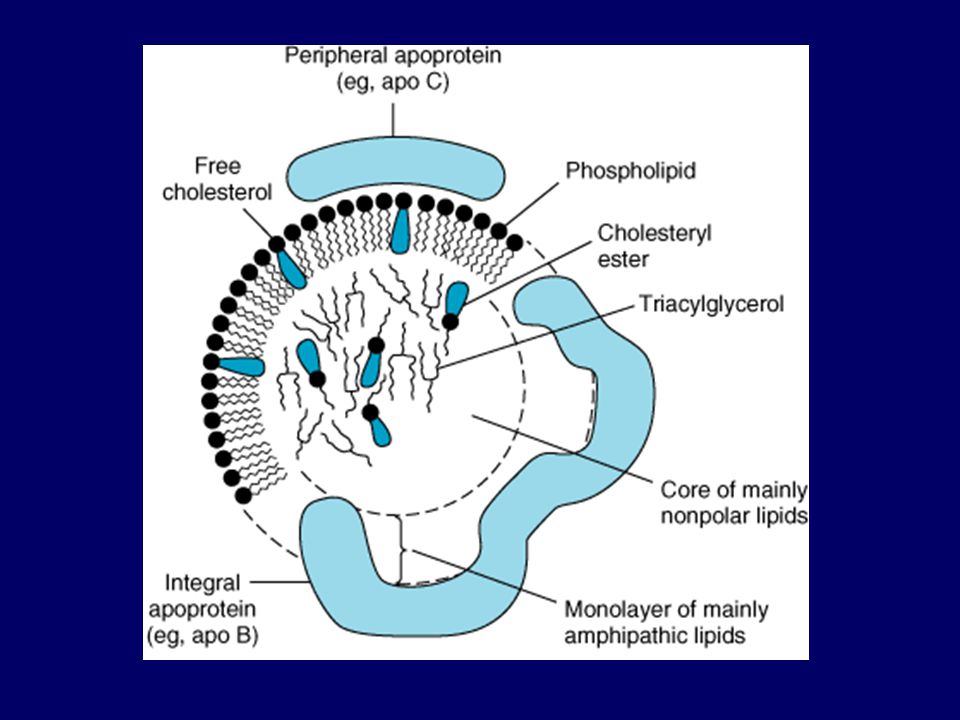
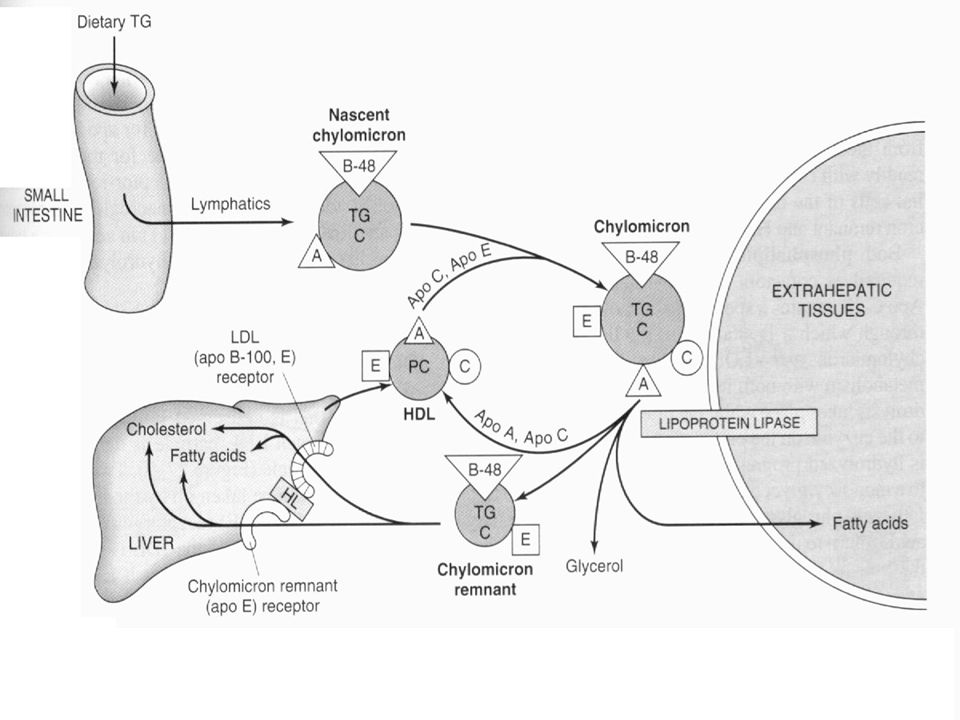
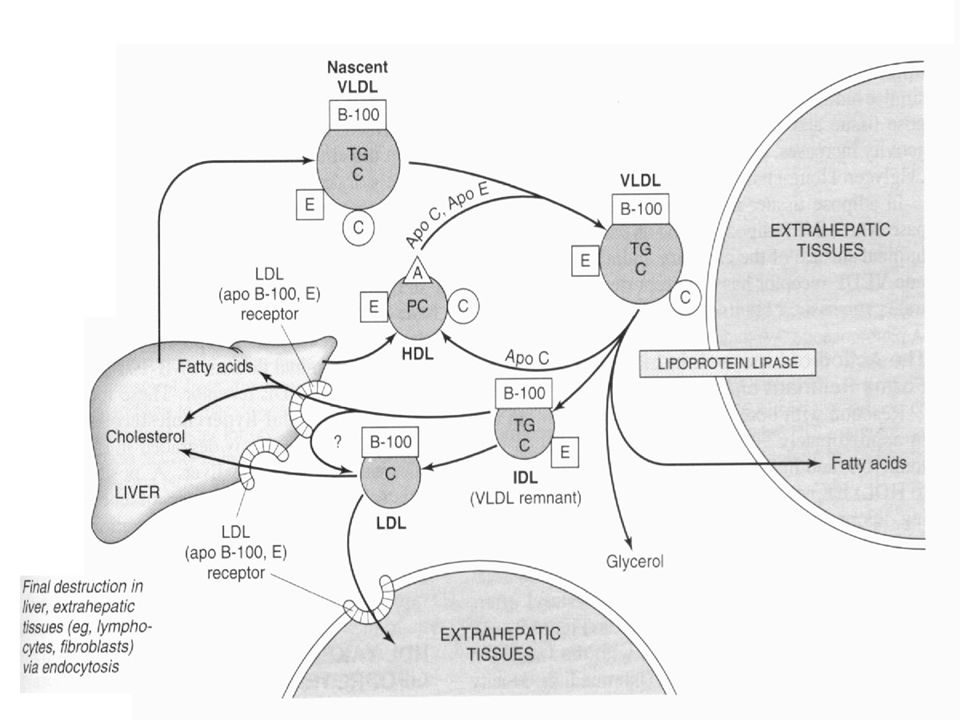

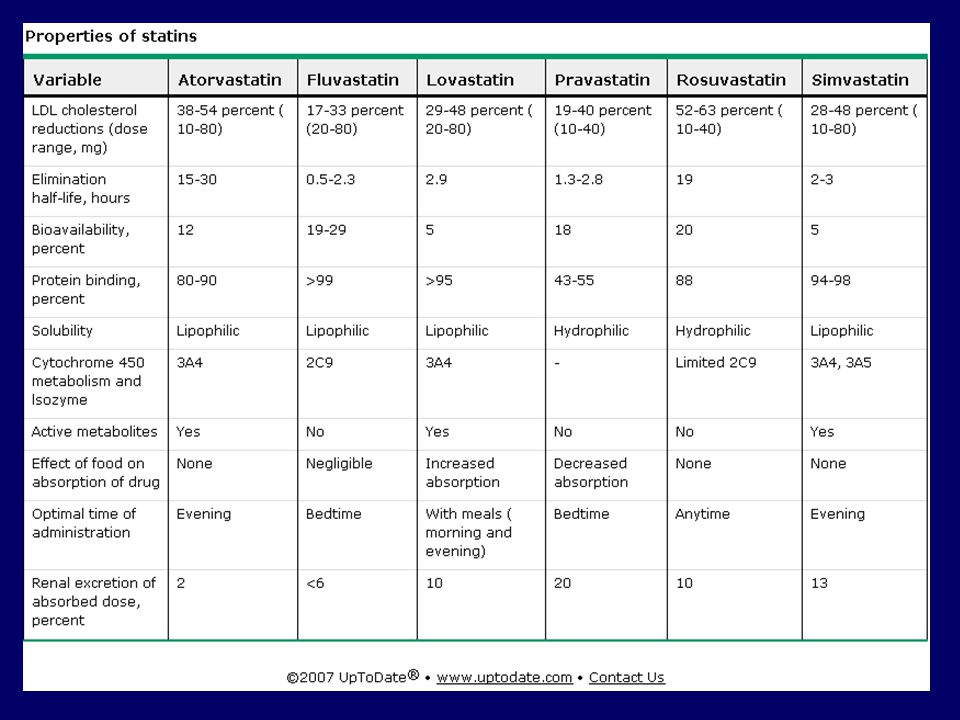
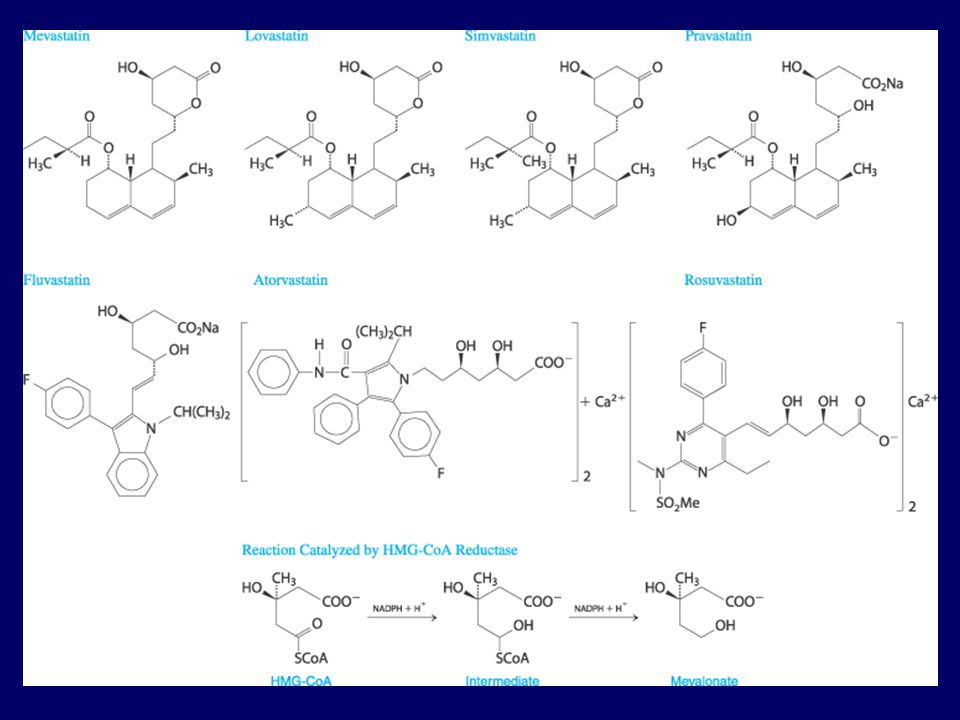

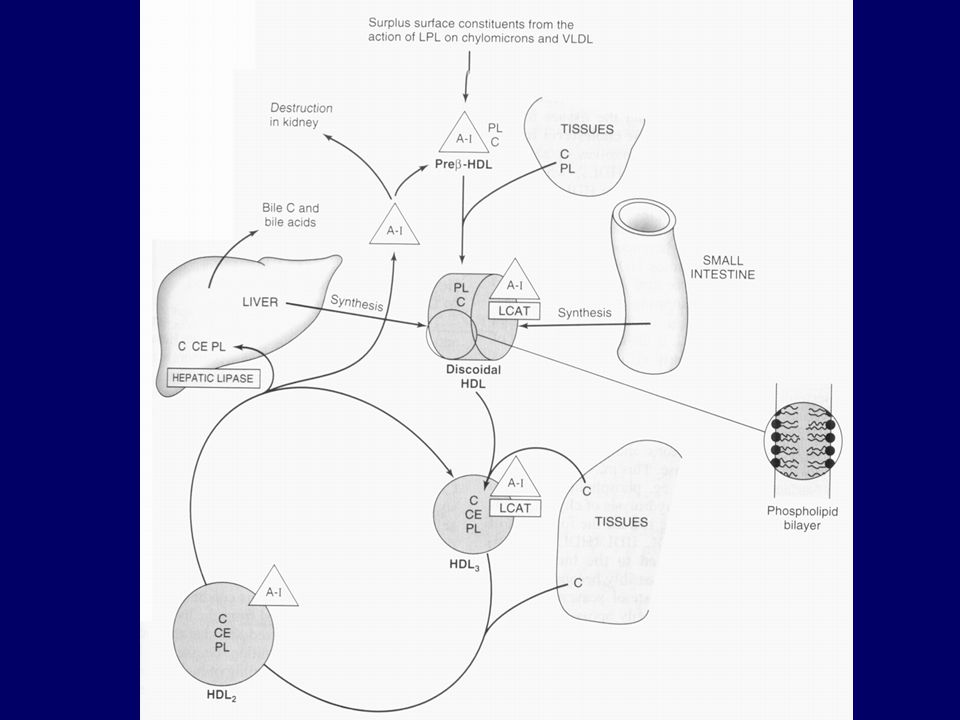
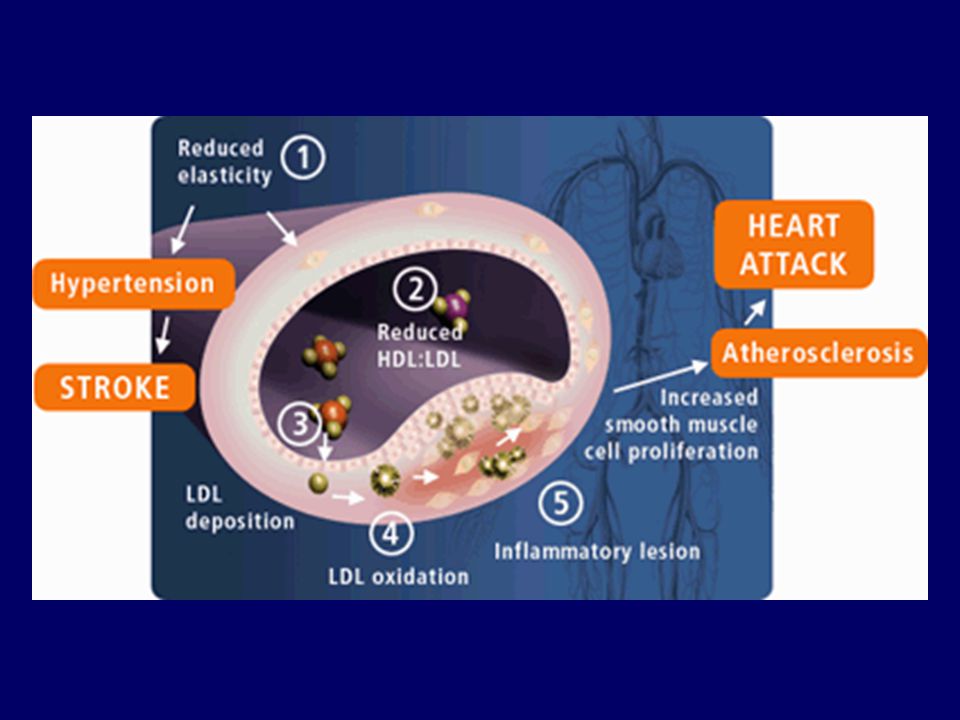
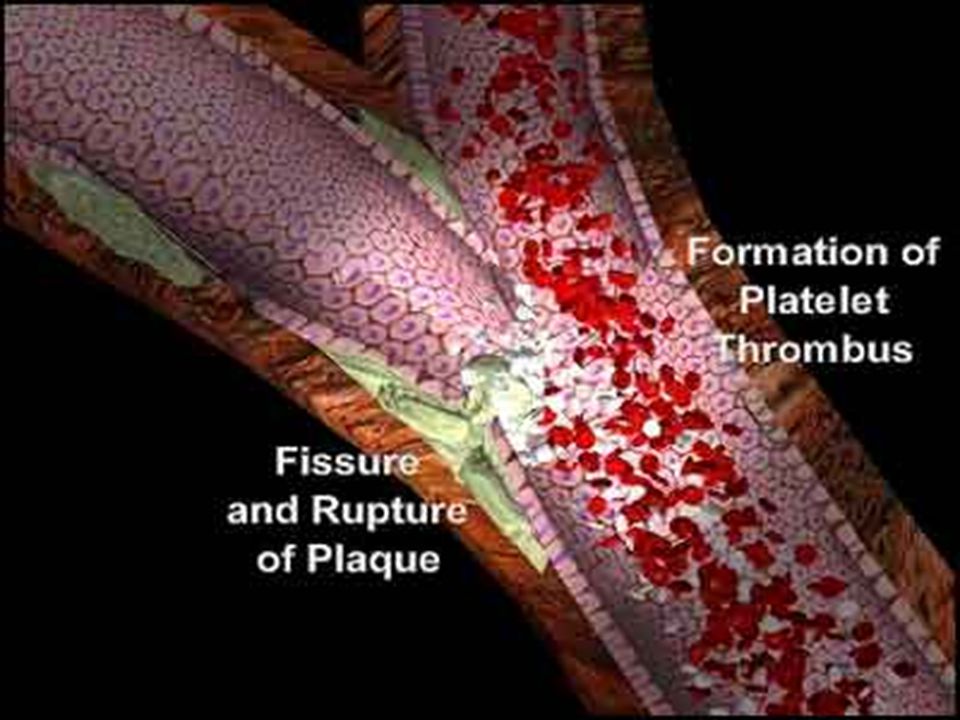
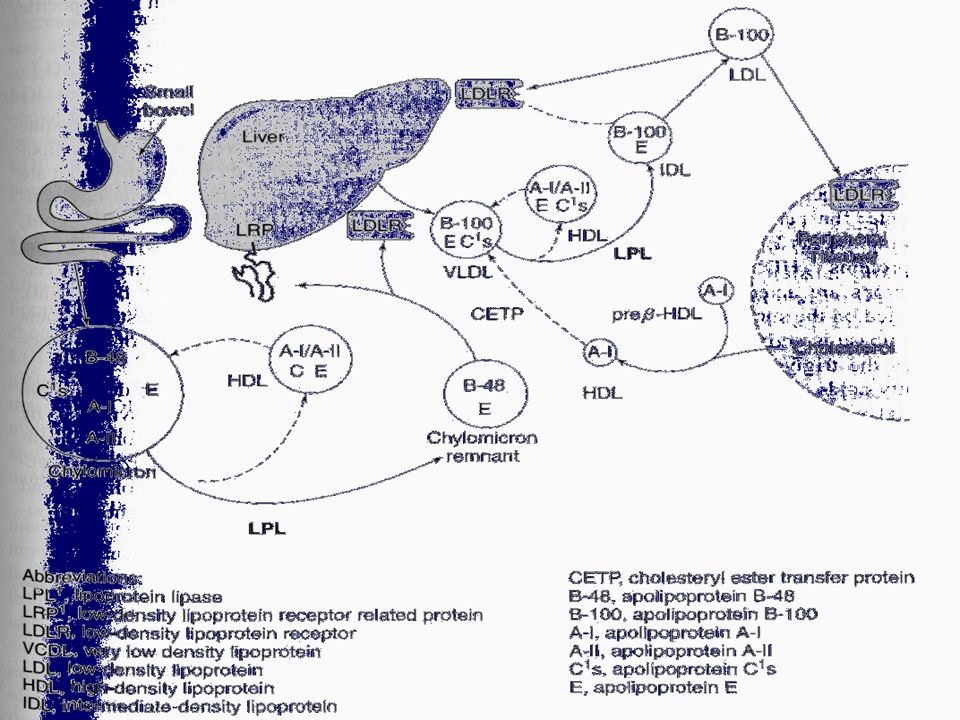
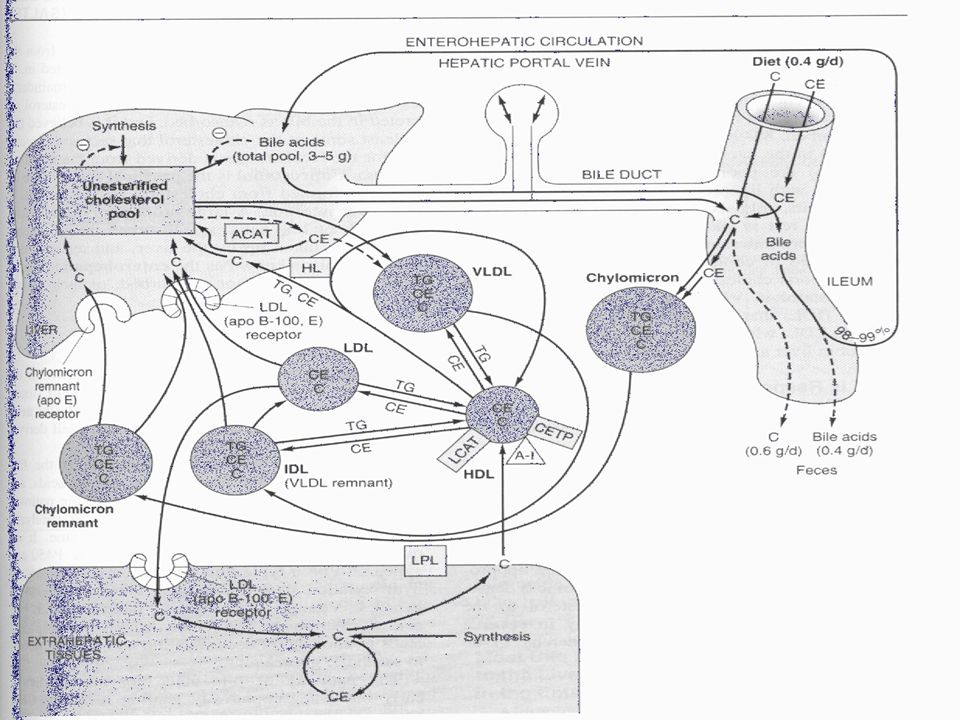

















>")

