Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
Anabilim Dalı Nöroloji Bilim Dalı Olgu Sunumu 17 Haziran 2014 Salı Uzm. Dr. Emek Uyur Yalçın

2
ÇOCUK NÖROLOJİSİ BİLİM DALI OLGU SUNUMU
17/06/2014 ÇOCUK NÖROLOJİSİ BİLİM DALI OLGU SUNUMU Dr. Emek Uyur Yalçın

3
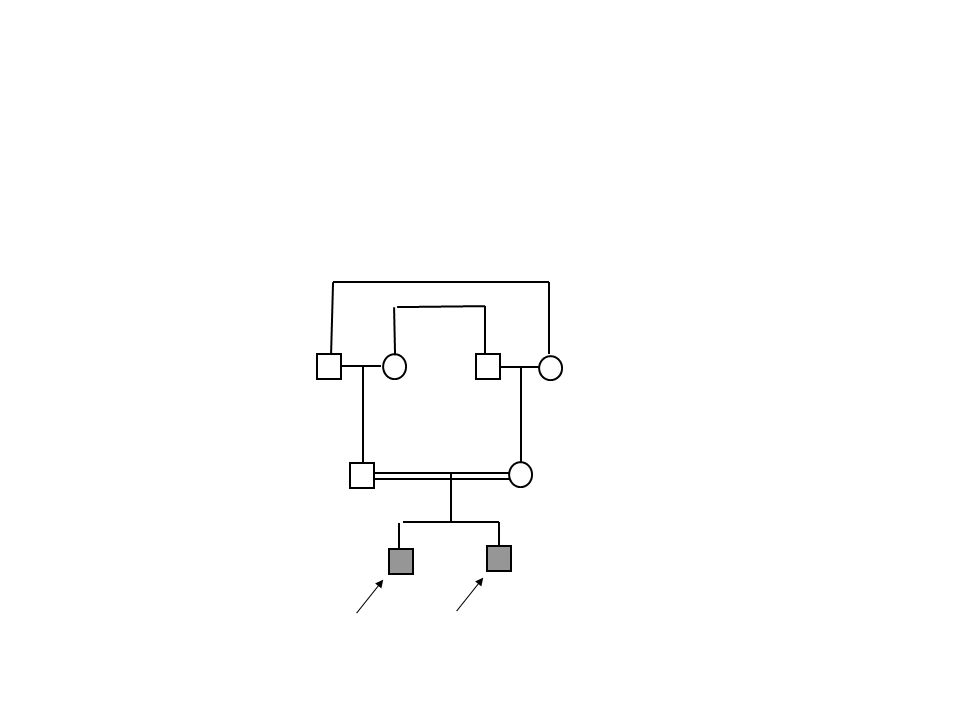
Olgu 1 8 aylık erkek hasta “Şikayet”
Başını tutamama, desteksiz oturamama “Öykü” Aile, hastanın 4 aylıkken başını tutamadığını, etrafla ilgisiz olduğunu fark ederek polikliniğe getirdi. “Özgeçmiş” Prenatal takibi düzensiz. G1P1A0 anneden, miadında, C/S ile 4200 gram olarak doğmuş. Postnatal adaptasyon sorunu olmamış. “Soygeçmiş” 1. derece kuzen evliliği

4
Olgu 1 ‘’Fizik Muayene’’
Mikrosefalik, göz takibi yok, başını tutamıyor, yaygın tonus artışı ve hiperaktif DTR’ler mevcuttu. ‘’Tetkikler’’ Hemogram, genişletilmiş biyokimya, Amonyak, laktat, Tiroid fonksiyon testleri, B12, folat, TANDEM-MS/MS, idrarda organik asit, Normal BOS ve serum kantitatif aminoasit incelemesi, BOS biyokimya ve mikroskobisi CDG taraması Göz muayenesi VEP, BAEP: İleti gecikmesi Karyotip analizi: 46,XY

5
Kranyal MRG: Diffüz serebral atrofi Korpus kallosumda incelme

7
Olgu 2: 37 haftalık, C/S ile, 2640 gram olarak doğmuş.
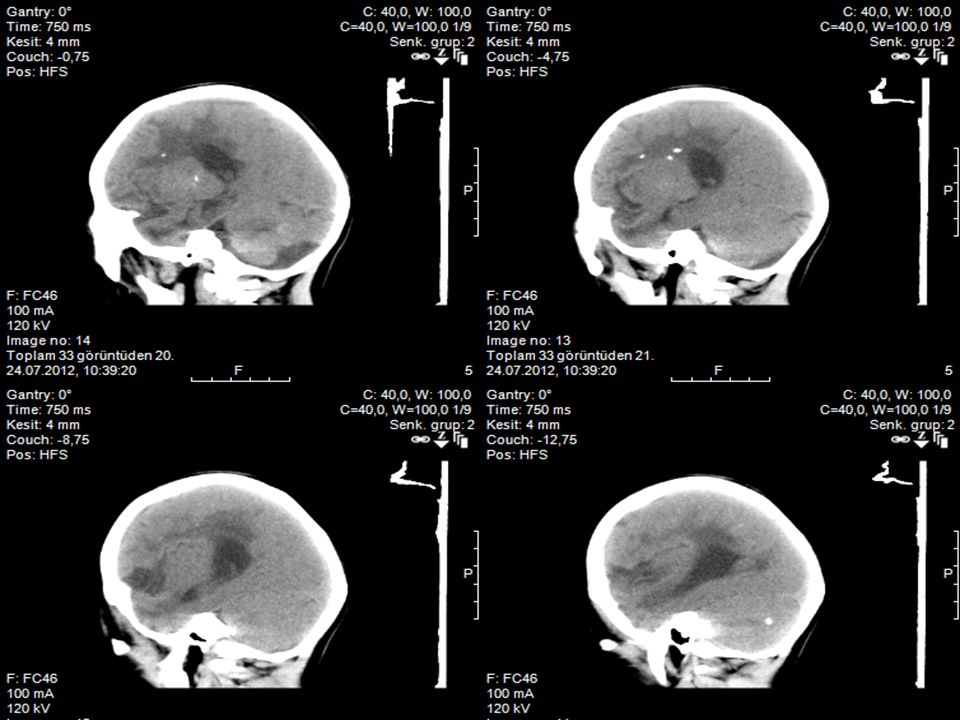
Prenatal dönemde oligohidramnioz nedeni ile takip edilmiş. Yenidoğan döneminde hepatosplenomegali, karaciğer enzimlerinde yükseklik, trombositopeni saptanmış. Konjenital CMV enfeksiyonu ön tanısı ile gansiklovir tedavisi verilmiş. Göz muayenesi ve işitme testi normal saptanmış. TFUS’da bilateral bazal gangliyonlarda ve beyin parankiminde multipl milimetrik kalsifikasyonlar Kraniyal BT’de periventriküler kalsifikasyon saptanmış.

8
2. Olgunun YD dönemindeki kranyal BT’si

9
1.Olgunun 1.5 yaşında çekilen kranyal BT’si

10
Olgu 3: 9 aylık erkek hasta “Şikayet”
Başını tam tutamama, desteksiz oturamama, etrafla ilgisiz olma “Öykü” Aile, hastanın yaşıtlarına göre gelişimimin geri olduğunu fark ederek polikliniğe getirdi. “Özgeçmiş” Prenatal takibi düzensiz. G5P4A0 anneden, miadında, C/S ile 2800 gram olarak doğmuş. Mekonyum aspirasyonu şüphesi nedeni ile 1 gün YDYBÜ’de takip edilmiş. “Soygeçmiş” 2. derece akraba evliliği Gelişme geriliği öyküsü olan ve 2 yaşında ex olan kız kardeş 10 yaşında epilepsi tanısı ile izlenen erkek kardeş

11
Olgu 3: ‘’Fizik muayene’’ mikrosefalik (<3. persentil)
göz takibi var, anneyi tanıyor, strabismus mevcut, baş traksiyonda geride kalıyor, yaygın tonus artışı ve hiperaktif DTR’ler mevcut, klonusu yok, diğer sistem muayeneleri doğal.

12
Olgu 3: Normal Hemogram, genişletilmiş biyokimya, Amonyak, laktat,
Tiroid fonksiyon testleri, B12, folat, TANDEM-MS/MS, İdrarda organik asit incelemesi Serum kantitatif aminoasit incelemesi, Göz muayenesi İşitme testi VEP: Ön görsel ileti yollarında gecikme Normal

13
Olgu 3: Kranyal MRG: Periventriküler derin beyaz cevherde diffüz T2 sinyal artışı, sol ventrikülde daha belirgin olmak üzere lateral ventriküllerde dilatasyon, korpus kallozumda incelme. Kranyal BT: Bilateral serebral hemisferde ve sol serebellumda parankimal kalsifikasyon

14
Olgu 3: 1.5 yaşında çekilen kranyal MRG

15
Olgu 3:

18
Ortak Patolojik Bulgular:
Mikrosefali Spastisite ve hiperaktif DTR’ler Global gelişme geriliği İntrakranyal kalsifikasyonlar Akraba evliliği ve ailede benzer klinik özelliklere sahip kardeş öyküsü

19
İntrakranyal Kalsifikasyon
Konjenital enfeksiyonlar (özellikle CMV) Neoplastik süreçler (tümör içi kalsifikasyonlar) Radyoterapi sonrası hasarlı beyin dokusunda izlenen kalsifikasyonlar (özellikle bazal gangliyonlarda) Distrofik kalsifikasyon (bakteriyel menenjit-ensefalit, hipoksik-iskemik hasar, iskemik stroke vb. süreçleri takiben serebral kortekste) Fokal kalsifikasyonlar (AV-malformasyon, kavernom vb.) Metastatik kalsifikasyonlar (hiper-, hipo- ve pseudohipoparatiroidizm, nefrojenik ve santral diabetes insipitus) Aicardi-Goutierés sendromu John H Livingston, Stavros Stivaros, Dan Warren, Yanick J Crow Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes Developmental Medicine & Child Neurology Volume 56, Issue 7 pages 612–626, July 2014
 Neoplastik süreçler (tümör içi kalsifikasyonlar) Radyoterapi sonrası hasarlı beyin dokusunda izlenen kalsifikasyonlar (özellikle bazal gangliyonlarda) Distrofik kalsifikasyon (bakteriyel menenjit-ensefalit, hipoksik-iskemik hasar, iskemik stroke vb. süreçleri takiben serebral kortekste) Fokal kalsifikasyonlar (AV-malformasyon, kavernom vb.) Metastatik kalsifikasyonlar (hiper-, hipo- ve pseudohipoparatiroidizm, nefrojenik ve santral diabetes insipitus) Aicardi-Goutierés sendromu. John H Livingston, Stavros Stivaros, Dan Warren, Yanick J Crow. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Developmental Medicine & Child Neurology Volume 56, Issue 7. pages 612–626, July")
20
Aicardi-Goutierés Sendromu
Olgu 1 ve olgu 2: RNASEH2C geninde c.196C>T (p.R66c) Anne ve baba taşıyıcı Olgu 3: TREX1 geninde c.341G>A (p.Arg114His)
 Anne ve baba taşıyıcı. Olgu 3: TREX1 geninde c.341G>A (p.Arg114His)")
21
Aicardi-Goutiéres Sendromu (AGS)
Pseudo-TORCH sendromu. Nadir görülen, sıklıkla erken çocukluk çağında başlangıç gösteren, beyin ve cildi etkileyen inflamatuar, progresif ve nörogelişimsel bir hastalıktır.

22
Tarihçe: BOS’ta artmış IFN-alfa seviyesi
1984: Jean Aicardi ve Françoise Goutieres tarafından 5 aileden 8 olgu bildirildi. erken başlangıçlı ağır ensefalopati mikrosefali serebral atrofi, BOS’ta kronik lenfositoz bazal gangliyon kalsifikasyonu serebral ak madde bozuklukları ölüm veya persistan vejetatif durum 1988: Pierre Lebon BOS’ta artmış IFN-alfa seviyesi

23
Yaklaşık 200 olgu, IAGSA (2010, Pavia/İtalya)
Bu olguların bir bölümünü; intrauterin enfeksiyon şüphesi olup, sonradan AGS tanı alan olgular oluşturmaktadır. Ancak bu olgularda, konjenital enfeksiyonu destekleyen seroloji pozitifliği saptanmamış. Elisa Fazzi , Marco Cattalini Simona Orcesi et al. Aicardi–Goutieres syndrome, a rare neurological disease in children: A new autoimmune disorder? Autoimmunity Reviews 12 (2013) 506–509
 506–509.")
24
Klinik Özellikler: Klinik özellikler geniş ve heterojen bir spektrum sergilemektedir. Erken süt çocukluğu döneminde hızla kaybedilen veya persistan vejetatif tabloya giren olgular ağırlıkta olmakla birlikte; minimal etkilenmiş olan (sadece ‘’chilblain’’ lezyonu olan ), normal eğitim alan ve daha stabil seyreden, daha uzun süre (4. dekada kadar) yaşayan olgular da bildirilmiş. Aynı aileden olan 2 kardeş olguda bile klinik tablo farklılık göstermektedir.
, normal eğitim alan ve daha stabil seyreden, daha uzun süre (4. dekada kadar) yaşayan olgular da bildirilmiş. Aynı aileden olan 2 kardeş olguda bile klinik tablo farklılık göstermektedir.")
25
Klinik Özellikler: Neonatal (erken başlangıçlı) Form:
Olguların %10-20’sini oluşturmakta. Jitterines, beslenme güçlüğü, neonatal nöbet, hipotoni, mikrosefali, intrauterin saptanmış serebral kalsifikasyon (hasarın intrauterin başladığını düşündüren), serebral atrofi Transaminazlarda artış, hepatosplenomegali, anemi, trombositopeni Konjenital enfeksiyonla ayırıcı tanı yapılmalı. Enfeksiyona yönelik kanıt yoksa; AGS’den şüphelenmek gerekir. Bu olguların 1/3’ü TREX-1 mutasyonu taşıyor, erken yaşta kaybediliyor.
, serebral atrofi. Transaminazlarda artış, hepatosplenomegali, anemi, trombositopeni. Konjenital enfeksiyonla ayırıcı tanı yapılmalı. Enfeksiyona yönelik kanıt yoksa; AGS’den şüphelenmek gerekir. Bu olguların 1/3’ü TREX-1 mutasyonu taşıyor, erken yaşta kaybediliyor.")
26
Klinik Özellikler: Geç Başlangıçlı Form:
Perinatal öyküde özellik olmayan ve gelişimi normale yakın seyreden olgularda; ortalama 3-4 ay civarında (1-2 yaş) hafif ensefalopati tablosuna benzeyen bir tablo ortaya çıkmakta. İrritabilite, uyku-uyanıklık siklusunun bozulması, beslenme güçlükleri, enfeksiyonun eşlik etmediği açıklanamayan hiperpireksi (erken alarm!). Nöromotor gelişimde duraklama, kazanılmış becerilerin kaybı, baş çevresinin büyümesinde yavaşlama, tonus değişiklikleri, distoni. Hastalığı tetikleyen, uyaran? Tablo neden aylar sonra ortaya çıkıyor?
 hafif ensefalopati tablosuna benzeyen bir tablo ortaya çıkmakta. İrritabilite, uyku-uyanıklık siklusunun bozulması, beslenme güçlükleri, enfeksiyonun eşlik etmediği açıklanamayan hiperpireksi (erken alarm!). Nöromotor gelişimde duraklama, kazanılmış becerilerin kaybı, baş çevresinin büyümesinde yavaşlama, tonus değişiklikleri, distoni. Hastalığı tetikleyen, uyaran Tablo neden aylar sonra ortaya çıkıyor")
27
Klinik Özellikler: Olguların bir kısmında doğumda glokom mevcut, bazılarında ise daha sonra gelişmiş. Bir bölümünde ise kortikal körlük mevcut. İşitme normal (konjenital viral enfeksiyonlardan ayırt etmede önemli!) Zamanla olguların %40’ından fazlasında ‘’chilblain’’ lezyonları gelişmiş. Sıklıkla el ve ayak parmaklarında, kulak kepçesinde görülen şiş ve kızarık lezyonlar ‘’Soğuk şişliği’’, soğuk maruziyeti ile kötüleşirler.
 Zamanla olguların %40’ından fazlasında ‘’chilblain’’ lezyonları gelişmiş. Sıklıkla el ve ayak parmaklarında, kulak kepçesinde görülen şiş ve kızarık lezyonlar. ‘’Soğuk şişliği’’, soğuk maruziyeti ile kötüleşirler.")
28
‘’Chilblain’’ lezyonu
A: RNASEH2B mutasyonu taşıyan Aicardi–Goutières tanılı 10 yaşında erkek olguda iskemik nekroza giden ayak parmakları. B: RNASEH2C mutasyonu taşıyan 3 yaşında kız olgunun ayak parmağında ‘’chilblain-benzeri’’ lezyon . Prendiville JS, Crow YJ. Blue (or purple) toes: chilblains or chilblain lupus-like lesions are a manifestation of Aicardi-Goutieres syndrome and familial chilblain lupus. J Am Acad Dermatol. 2009 Oct;61(4):727-8.
 toes: chilblains or chilblain lupus-like lesions are a manifestation of Aicardi-Goutieres syndrome and familial chilblain lupus. J Am Acad Dermatol Oct;61(4):")
29
Tanı: Laboratuvar: Metabolik ve enfeksiyöz tarama normal.
BOS’ta hücre artışı (özellikle lenfositoz) BOS’ta interferon-alfa ve neopterin artışı Nöroradyoloji: Tipik olarak, bilateral bazal gangliyonlarda ve ak maddede kalsifikasyonlar Özellikle frontal ve temporal bölgelerde, kistik dejenerasyon özelliği gösterebilen ak madde anormallikleri (%75-100) Serebral atrofi
 BOS’ta interferon-alfa ve neopterin artışı. Nöroradyoloji: Tipik olarak, bilateral bazal gangliyonlarda ve ak maddede kalsifikasyonlar. Özellikle frontal ve temporal bölgelerde, kistik dejenerasyon özelliği gösterebilen ak madde anormallikleri (%75-100) Serebral atrofi.")
30
Genetik Olguların %90’ından 7 gen sorumlu.
Type OMIM Gene Locus AGS1 225750 TREX1 3p21.31 AGS2 610181 RNASEH2B 13q14.3 AGS3 610329 RNASEH2C 11q13.1 AGS4 610333 RNASEH2A 19p13.2 AGS5 612952 SAMHD1 20q11.23 AGS6 615010 ADAR 1q21.3 AGS7 60695 IFIH1 2q24 Olguların %90’ından 7 gen sorumlu. OR kalıtım, nadir olarak OD kalıtım ve ‘‘de novo’’ mutasyon.

31
Genetik Bu genler; nükleik asit tamir mekanizmasında önemli rol oynamakta ve adı geçen genlerdeki mutasyonlar otoimmun süreçlerin sekonder aktivasyonuna yol açmaktadır.

32
İzlemde; Olgu 1 Olgu 2 6 yaşında
2 yaşında nöbetleri başladı, VPA alırken nöbetleri kontrol altında Mikrosefalik Trunkal hipotoni, ekstremitelerde hipertoni Baş kontrolü yok, göz takibi var FTR desteği alıyor 2.5 yaşında 2 yaşında glokom saptandı. Sık alt solunum yolu enfeksiyonu öyküsü var GER nedeniyle opere oldu, gastrostomili Mikrosefalik, baş kontrolü yok, hipoton Nöbeti yok FTR desteği alıyor

33
İzlemde; Olgu 3: 3.5 yaşında Mikrosefalik
Baş kontrolü yok, göz takibi kısa süreli, anneyi tanıyor, FTR desteği alıyor, Baklofen tedavisi alıyor, Nöbeti yok.

34
Aicardi-Goutierés Sendromunu düşün!
Son Söz; Konjenital enfeksiyonları taklit eden nöromotor gelişme geriliği, mikrosefali, intrakranyal kalsifikasyon vb. klinik ve radyolojik bulgular ile karşılaşıldığında; serolojik testler negatif ise, işitme normalse, BOS’ta enfeksiyonun eşlik etmediği lenfositoz varsa, BOS’ta IFN-alfa yüksek ise, ailede benzer özellik gösteren olgu varsa; Aicardi-Goutierés Sendromunu düşün!

35
Son Söz; Etiyolojisi net olarak aydınlatılamayan nörolojik hastalıklarda, kranyal MRG ile birlikte Kranyal BT yol gösterici olabilir!

36
Teşekkürler…

Benzer bir sunumlar