Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
Amino Asit Hastalıkları

3
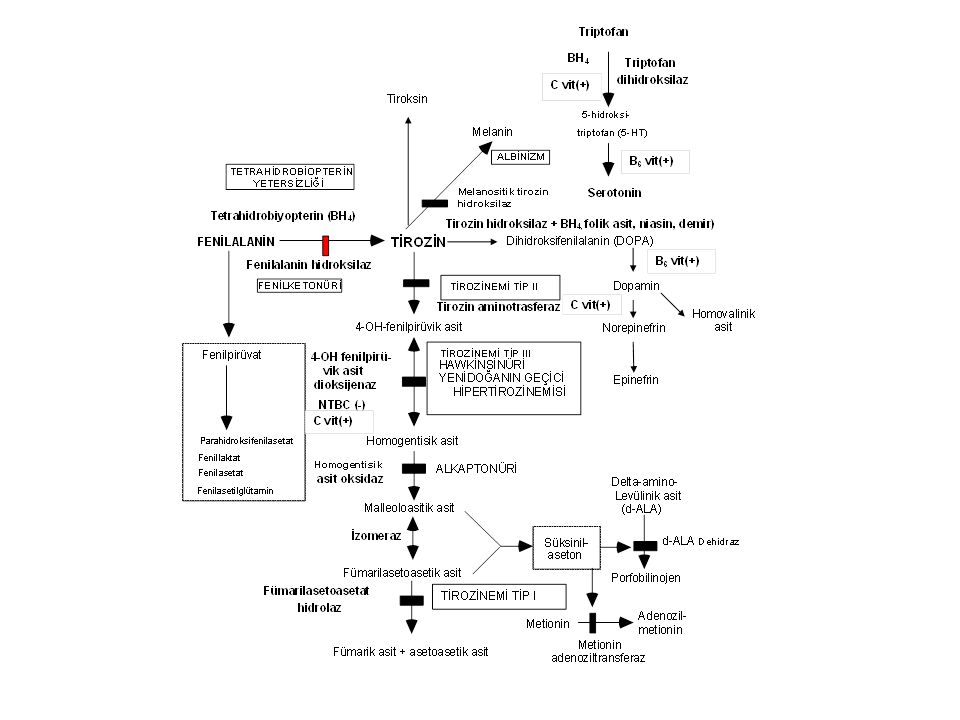
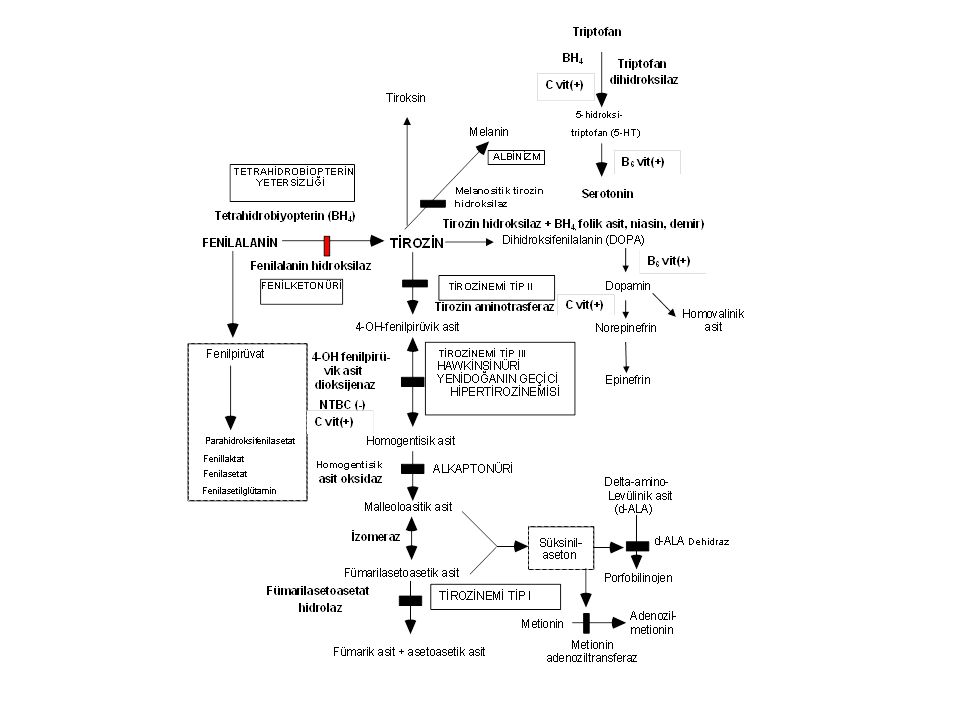
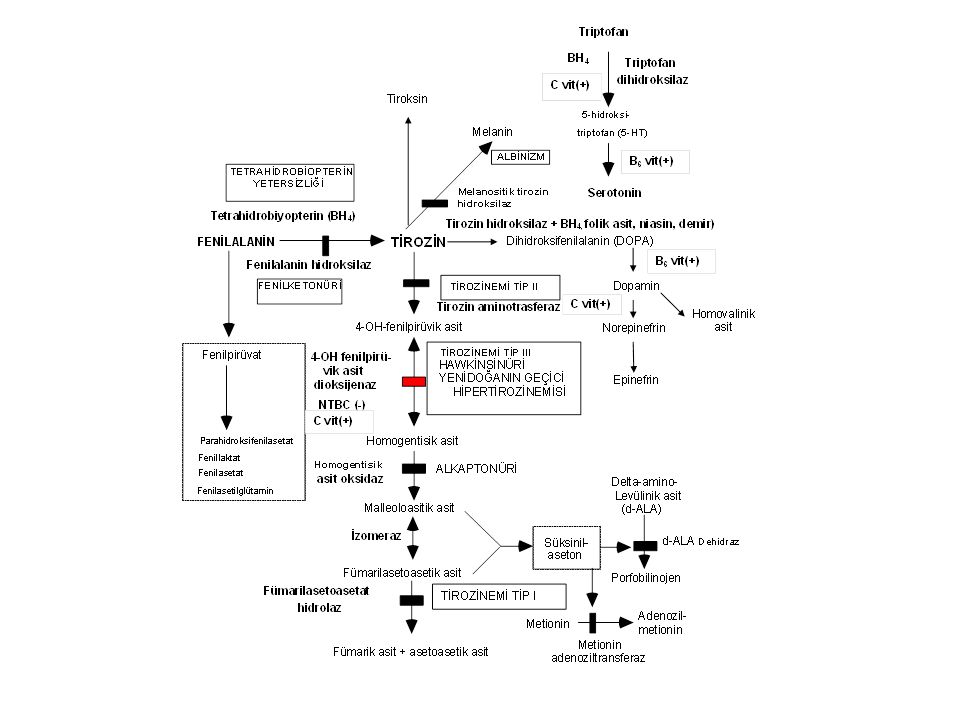
Fenilketonüri (PKU) Enzim defekti: Fenilalanin hidroksilaz(12q22-q24.1): 400'den fazla mutasyon vardır. İnsidans: Ortalama 1:10,000’dır; en yüksek insidans Türkiye'dedir (1: 4,000).
.")
4
Fenilketonüri sınıflama
1. Klasik Fenilketonüri (tam ya da tama yakın enzim yetersizliği) Kan fenilalanin >20 mg/dL (>1200 mmol/L). Rijit diyet gerektirir 2. Atipik (Hafif) Fenilketonüri (enzim aktivitesi %1-5) Kan Fenilalanin <10-20mg/dL. Kısmi diyet gerektirir 3. Selim Fenilketonüri (enzim aktivitesi >%5) Kan fenilalanin <10 mg/dL; bulgu yok, diyet gerektirmez. 4. Habis fenilketonüri A. Tetrahidrobiopterin sentez yetersizlikleri B. Tetrahidrobiopterin siklusu defektleri
 Kan fenilalanin >20 mg/dL (>1200 mmol/L). Rijit diyet gerektirir. 2. Atipik (Hafif) Fenilketonüri (enzim aktivitesi %1-5) Kan Fenilalanin <10-20mg/dL. Kısmi diyet gerektirir. 3. Selim Fenilketonüri (enzim aktivitesi >%5) Kan fenilalanin <10 mg/dL; bulgu yok, diyet gerektirmez. 4. Habis fenilketonüri. A. Tetrahidrobiopterin sentez yetersizlikleri. B. Tetrahidrobiopterin siklusu defektleri.")
5
Fenilketonüri- Klinik bulgular
Ağır beyin hasarı, ilerleyici motor-mental gerilik Spastisite, felçler Otizm, hiperaktivite Konvülsiyon, self-mütilasyon Açık saç ve deri rengi (sarı saç, mavi göz, tirozin yetersizliği), skleroderma İdrarda ve terde fare leşi kokusu (fenilasetat)
, skleroderma. İdrarda ve terde fare leşi kokusu (fenilasetat)")
7
Fenilketonüri-Tanı Kan fenilalanin düzeyinin 20 mg/dL'den yüksek olması Plazma tirozin düzeyinin normal/düşük olması (Phe/Tyr >3) İdrarda fenilalanin metabolitlerinin artması Kofaktör tetrahidrobiopterin metabolizmasını normal olması (BH4 yükleme testi)
")
8
Fenilketonüri-Tedavi
Fenilalanin >12-15mg/dL ise fenilalaninsiz ya da fenilalaninden fakir diyet Tedavinin hedefi 0-10yıl: fenilalanin değerleri: mg/dL 11-16yıl: fenilalanin değerleri: <15 mg/dL <16yıl: fenilalanin değerleri: <20 mg/dL PKU’lu gebe kadınlar: fenilalanin değerleri < 7mg/dL

9
Maternal fenilketonüri = feniketonürik fetopati
Normal fenilalanin düzeyi Mikrosefali Kardiyak defektler Motor-mental retardasyon

11
HABİS FENİLKETONÜRİ 1. Tetrahidrobiopterin sentez yetersizlikleri: Guanozin trifosfat siklohidrolaz I (GTPCH) ve 6-piruvoil-tetrahidrobiopterin sentaz (PTPS) yetersizlikleri 2. Tetrahidrobiopterin siklusu defektleri: Dihidropteridin redüktaz (DHPR) ve pteridin 4 alfa-karbinolamin dehidraz (PCD) yetersizlikleri. BH4 yetersizliği dopamin ve serotonin sentezini bozar. Hastalık kendisini konvülsiyon, hipotoni, koreiform hareketler ve psikomotor gerilik ile ortaya koyar. Tetrahidrobiyopterin yükleme testi yapılırsa fenilalanin düzeyleri birkaç saat içinde düşer. Tedavi: Diyet tek başına yetersizdir. Ek olarak BH4 + L-dopa + 5-hidroksitriptofan verilir.
 ve 6-piruvoil-tetrahidrobiopterin sentaz (PTPS) yetersizlikleri. 2. Tetrahidrobiopterin siklusu defektleri: Dihidropteridin redüktaz (DHPR) ve pteridin 4 alfa-karbinolamin dehidraz (PCD) yetersizlikleri. BH4 yetersizliği dopamin ve serotonin sentezini bozar. Hastalık kendisini konvülsiyon, hipotoni, koreiform hareketler ve psikomotor gerilik ile ortaya koyar. Tetrahidrobiyopterin yükleme testi yapılırsa fenilalanin düzeyleri birkaç saat içinde düşer. Tedavi: Diyet tek başına yetersizdir. Ek olarak BH4 + L-dopa + 5-hidroksitriptofan verilir.")
13
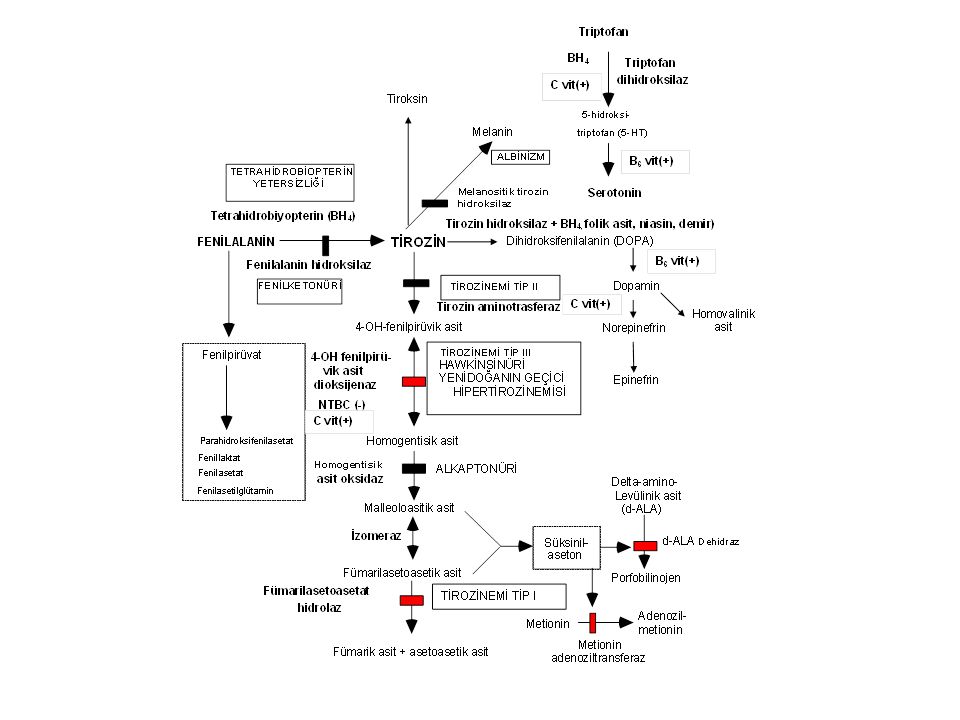
Tirozinemi Tip I- Klinik bulgular
Enzim defekti: Fumarilasetoasetat hidroksilaz Klinik bulgular Akut infantil form: Ağır karaciğer yetersizliği, kusma, kanamalar, sepsis, renal tubulopati (Fanconi sendromu) Kronik form: Hepatomegali, siroz, büyüme geriliği, rahitis, hematom, tübülopati, periferik nöropati ve karın ağrıları (d-ALA artışı)
 Kronik form: Hepatomegali, siroz, büyüme geriliği, rahitis, hematom, tübülopati, periferik nöropati ve karın ağrıları (d-ALA artışı)")
14
Tirozinemi Tip I-Tanı Yüksek süksinilaseton düzeyi (diagnostik).
Tirozin: Normal ya da yüksek. Metionin: yüksek Delta-aminolevulinik asit: yüksek Alfa-feto protein: çok yüksek (hepatosellüler karsinom belirteci)
")
15
Tirozinemi Tip I-Tedavi
Tedavi: NTBC 1 mg/kg: toksik metabolitlerin (süksinilaseton) birikimini engeller; Bu sırada tirozin daha da yükseldiği için tirozinden kısılı diyet daha da önem taşır. İlaç tedavisi başarılı olmazsa karaciğer transplantasyonu
 birikimini engeller; Bu sırada tirozin daha da yükseldiği için tirozinden kısılı diyet daha da önem taşır. İlaç tedavisi başarılı olmazsa karaciğer transplantasyonu.")
16
Tirozinemi Tip I-Komplikasyonlar
Komplikasyonlar: Karaciğer yetersizliği, hepatosellüler karsinom (alfa-feto protein takibi) Prognoz: NTBC tedavisi ile oldukça iyi. Aksi halde karaciğer transplantasyonu yapılır.
 Prognoz: NTBC tedavisi ile oldukça iyi. Aksi halde karaciğer transplantasyonu yapılır.")
18
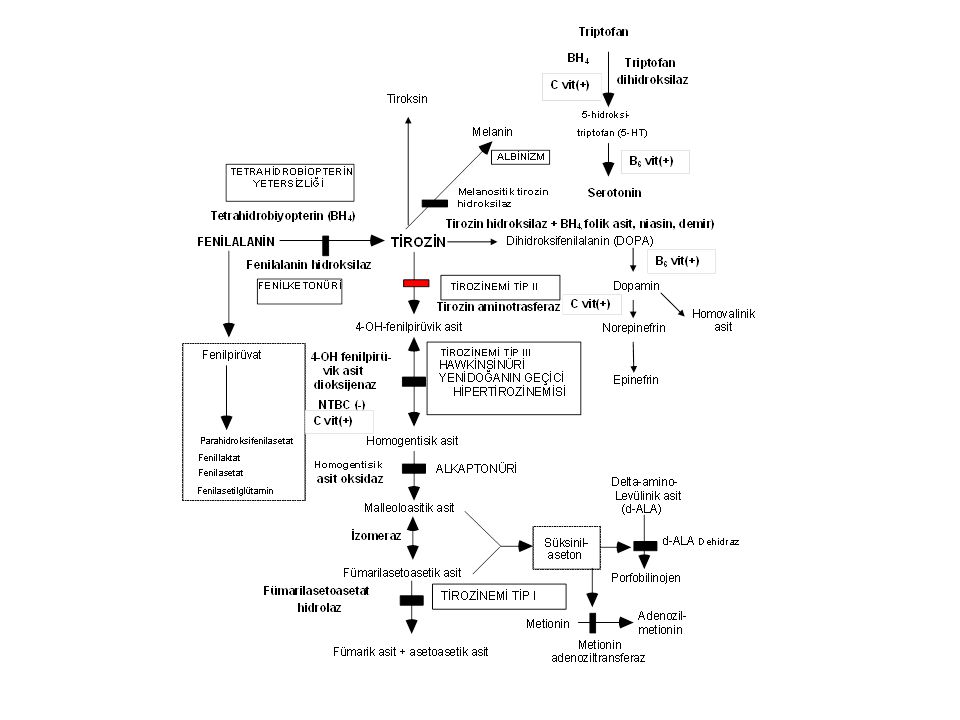
Tirozinemi Tip II Enzim defekti: Sitozolik tirozin aminotransferaz
Klinik bulgular: Ağrılı kornea lezyonları (yaşarma, fotofobi, sikatris), hafif mental gerilik. Tanı: Tirozin ve fenilalanin düzeyinde yük-seklik Tedavi: Tirozin ve fenilalaninden fakir diyet
, hafif mental gerilik. Tanı: Tirozin ve fenilalanin düzeyinde yük-seklik. Tedavi: Tirozin ve fenilalaninden fakir diyet.")
20
Yenidoğanın geçici hipertirozinemisi
pHPPD immatüritesi sonucu gelişir. Normal yenidoğanlarda (1:10) ve prematür çocuklarda (1:3) iki hafta kadar sürer. Kalıcı bir hasara neden olmaz. C vitamini ve diyet ayarlamasına iyi cevap verir.
 ve prematür çocuklarda (1:3) iki hafta kadar sürer. Kalıcı bir hasara neden olmaz. C vitamini ve diyet ayarlamasına iyi cevap verir.")
21
Alkaptonüri Enzim defekti: Homogentisik asit oksijenaz
Klinik bulgular: İdrarın asit ortamda ya da bekletilince siyahlaşması, erişkinlerde ağır artrit Tanı: İdrarda homogentisik asit artışı Tedavi: Proteinden kısılı diyet? NTBC? Prognoz: Geç yaşlarda ağır artroz görülebilir.

22
Kükürtlü aminoasitlerin metabolizması

23
Homosistinüri-Klinik belirtiler
Lens dislokasyonu (genellikle aşağı doğru), glokom, retina ayrılması, miyopi, katarakt Araknodaktili, hipermobilite, pektus ekskavatum, düz tabanlık, dolikosternomeli, osteoporoz, genu valgum, diş bozuklukları Zekâ genellikle geri, konvülsiyonlar, EEG bozuklukları, spastisite, fokal nörolojik bozukluklar, psikiatrik bozukluklar Arteriyel ve venöz trombuslar, yüz kızarması, livedo reticularis Genel anestezi: Kontrendike Ayırıcı tanı: Marfan sendromu
, glokom, retina ayrılması, miyopi, katarakt. Araknodaktili, hipermobilite, pektus ekskavatum, düz tabanlık, dolikosternomeli, osteoporoz, genu valgum, diş bozuklukları. Zekâ genellikle geri, konvülsiyonlar, EEG bozuklukları, spastisite, fokal nörolojik bozukluklar, psikiatrik bozukluklar. Arteriyel ve venöz trombuslar, yüz kızarması, livedo reticularis. Genel anestezi: Kontrendike. Ayırıcı tanı: Marfan sendromu.")
24
Marfan sendromu

25
Homosistinüri-Tanı-Tedavi
Taze idrarda siyanid nitroprussid testinin pozitif olması Kanda homosistin ve metionin artmış sistin azalmıştır. Farmakolojik dozlarda ( mg/gün) piridoksin verilen, residüel enzim aktivitesini arttırır. Tedavi yetersiz olursa diyet + betain 100 mg/kg Metionin'den fakir, sistinden zengin diyet Hedef homosistein düzeyinin 30µmol/L’nin altında tutulmasıdır.
 piridoksin verilen, residüel enzim aktivitesini arttırır. Tedavi yetersiz olursa diyet + betain 100 mg/kg. Metionin den fakir, sistinden zengin diyet. Hedef homosistein düzeyinin 30µmol/L’nin altında tutulmasıdır.")
26
Maternal hiperhomosisteinemi
Maternal hiperhomosisteinemi: konjenital defektler Nöral tüp defektleri Kardiak çıkış defektleri? Dudak-damak yarıkları? Renal defektler? Pilor stenozu? Down sendromu Tanı: Yüksek (30-40µmol/L’ye kadar) homosistein düzeyleri Tedavi: Folik asit 5mg/gün, B6 Vitamini: 100 mg/gün Korunma: Folik asitten zengin ya da zenginleştirilmiş gıdalar verilir.
 homosistein düzeyleri. Tedavi: Folik asit 5mg/gün, B6 Vitamini: 100 mg/gün. Korunma: Folik asitten zengin ya da zenginleştirilmiş gıdalar verilir.")
27
Hafif hiperhomosisteinemi
Nedenler Metilen tetrahidrofolat redüktaz (MTHFR) polimorfizmi, termolabil varyant, %5 ile %60 arasında değişen homozigotluk oranı Sistationin-ß-sentaz heterozigotluğu Folik asit metabolizmasının endojen ve ekzojen bozuklukları Vitamin B12 ve B6 yetersizlikleri Klinik bulgular 3ncü ve 4ncü on yılda erken vasküler hastalık (enfarktlar, trombozlar, tromboemboliler)
 polimorfizmi, termolabil varyant, %5 ile %60 arasında değişen homozigotluk oranı. Sistationin-ß-sentaz heterozigotluğu. Folik asit metabolizmasının endojen ve ekzojen bozuklukları. Vitamin B12 ve B6 yetersizlikleri. Klinik bulgular. 3ncü ve 4ncü on yılda erken vasküler hastalık (enfarktlar, trombozlar, tromboemboliler)")
28
NONKETOTİK HİPERGLİSİNEMİ
Doğumda ya da doğumdan sonraki birkaç gün içinde sonra perina-tal hipoksisi olmayan bir bebekte koma, hipotoni, hıçkırık ve konvülsiyonlar (özellikle miyoklonik sıçramalar) görülür. Hasta sürekli uyur. Hastalık genellikle ölümcüldür. EEG'deki tipik patlama dalgaları olguların hepsinde mevcuttur. Hastalığın hafif tipleri zekâ geriliği, spastisite ve konvülsiyonlar ile karakterizedir. BOS:serum glisin oranı yüksek Tedavide kullanılan striknin (glisin reseptör antagonisti), sodyum benzoat (glisini tutar), metionin, piridoksin ve lökovorin ile alınan sonuçlar başarılı değildir
 görülür. Hasta sürekli uyur. Hastalık genellikle ölümcüldür. EEG deki tipik patlama dalgaları olguların hepsinde mevcuttur. Hastalığın hafif tipleri zekâ geriliği, spastisite ve konvülsiyonlar ile karakterizedir. BOS:serum glisin oranı yüksek. Tedavide kullanılan striknin (glisin reseptör antagonisti), sodyum benzoat (glisini tutar), metionin, piridoksin ve lökovorin ile alınan sonuçlar başarılı değildir.")
29
Üre döngüsü defektleri

30
Protein Amonyak Üre döngüsü Üre

31
Carbaglu (+)
")
32
Üre döngüsü defektleri: Etyoloji ve Genetik (Kümülatif insidans: 1:10,000)
Ornitin transkarbamilaz yetersizliği (en sık görülen defekt, X-e bağlı) Arjininosüksinat sentaz yetersizliği =sitrullinemi (OR) Karbamilfosfat sentaz yetersizliği (OR) Arjininosuksinat liaz yetersizliği =Arjininosuksinik asidüri (OR) Arjinase yetersizliği = arjininemi (OR) N-asetil glütamat sentaz yetersizliği (OR)
 Arjininosüksinat sentaz yetersizliği =sitrullinemi (OR) Karbamilfosfat sentaz yetersizliği (OR) Arjininosuksinat liaz yetersizliği =Arjininosuksinik asidüri (OR) Arjinase yetersizliği = arjininemi (OR) N-asetil glütamat sentaz yetersizliği (OR)")
33
Üre döngüsü defektleri: Klinik belirtiler
Hipo- hipertermi Hipervantilasyon (respiratuvar alkaloz) Emme bozukluğu Letarji Refleks kaybı İntrakraniyal kanama Nöbetler İlerleyici ansefalopati
 Emme bozukluğu. Letarji. Refleks kaybı. İntrakraniyal kanama. Nöbetler. İlerleyici ansefalopati.")
34
Üre döngüsü defektleri: Ayırıcı tanı
Organik asidüriler: Çok karışır, metabolik asidozun olmaması ile ayırdedilir. Karaciğer hastalıkları: neonatal hepatit, galaktozemi, tirozinemi, solunum zinciri hastalıkları Yenidoğanın geçici hiperammonemisi: patent ductus venosus’a bağlı.

35
CPS= Karbamoil fosfat sentaz OTC= Ornitin transkarbomoilaz ASA=Arjininosüksinik asit AS=Arjininosüksinat sentaz AL=Arjininosüksinat liaz(sitrüllinemi)
")
36
Üre döngüsü defektleri: tedavi
Protein alımının durdulması Katabolizmanın azaltılması: yüksek kalorili enfüzyon (karbohidrat + lipid) Amonyağın mekanik yoldan uzaklaştırılması (>400 µmol/L) : hemodiyafiltrasyon, hemofiltrasyon, ya da hemodiyaliz (periton dializi çok etkili değil) Arjinin: mg/kg/gün (üre siklusunu destekler) Sodyum benzoat: 350mg/kg/gün Sodyum fenilbütirat: 250mg/kg/gün Karglumik asit (Carbaglu®): mg/kg/gün (NAGS yetersizliği) (üre siklusunu destekler)
 Amonyağın mekanik yoldan uzaklaştırılması (>400 µmol/L) : hemodiyafiltrasyon, hemofiltrasyon, ya da hemodiyaliz (periton dializi çok etkili değil) Arjinin: mg/kg/gün (üre siklusunu destekler) Sodyum benzoat: 350mg/kg/gün. Sodyum fenilbütirat: 250mg/kg/gün. Karglumik asit (Carbaglu®): mg/kg/gün (NAGS yetersizliği) (üre siklusunu destekler)")
37
Carbaglu (+)
")
38
Üre döngüsü defektleri: tedavi
Arjinin: mg/kg/gün (üre siklusunu destekler) Sodyum benzoat: 350mg/kg/gün Sodyum fenilbütirat: 250mg/kg/gün Karglumik asit (Carbaglu®): mg/kg/gün (NAGS yetersizliği) (üre siklusunu destekler)
 Sodyum benzoat: 350mg/kg/gün. Sodyum fenilbütirat: 250mg/kg/gün. Karglumik asit (Carbaglu®): mg/kg/gün (NAGS yetersizliği) (üre siklusunu destekler)")
39
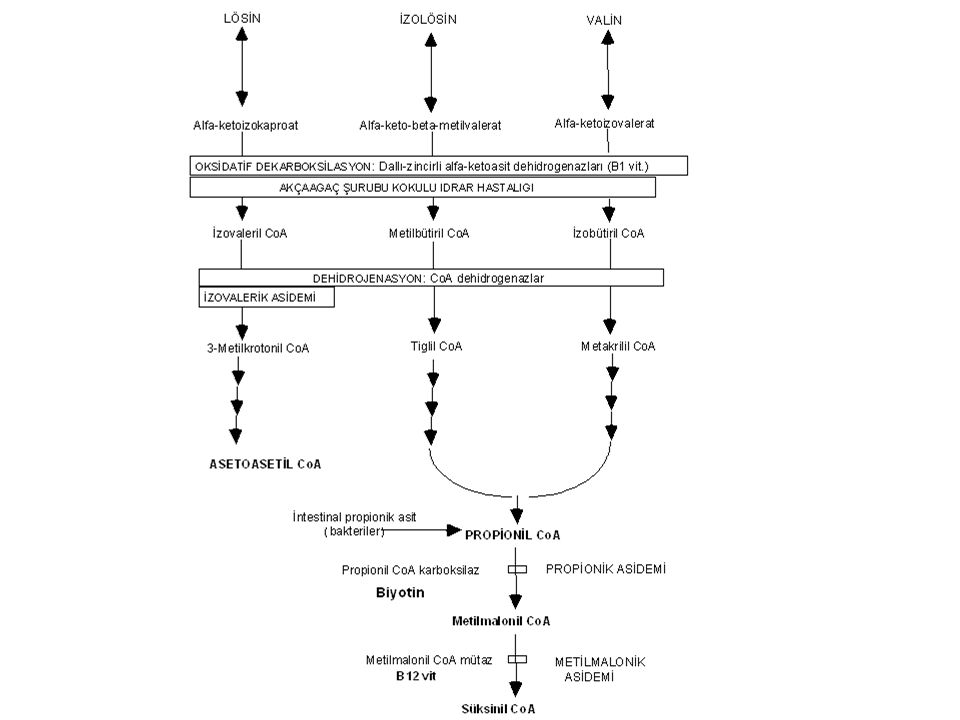
Organik asidemiler

40
Normal elektrolit dağılımı
(Katyonlar= Anyonlar) Sodyum (140 mmol/L)+ Potasyum (5 mmol/L) = Bikarbonat (25 mmol/L)+ klor (100 mmol/L) + Organik asitler (12-20 mmol/L) Anyon açığı = Ölçülmeyen organik asitler (Sodyum + Potasyum)– (Bikarbonat + Klor)= <12-20 mmol/L
 Sodyum (140 mmol/L)+ Potasyum (5 mmol/L) = Bikarbonat (25 mmol/L)+ klor (100 mmol/L) + Organik asitler (12-20 mmol/L) Anyon açığı = Ölçülmeyen organik asitler. (Sodyum + Potasyum)– (Bikarbonat + Klor)= <12-20 mmol/L.")
41
Anyon açığının arttığı(normokloremik) bir metabolik asidoz örneği
Sodyum (140 mmol/L) Potasyum (5 mmol/L) Bikarbonat (10 mmol/L) Klor (100 mmol/L) (140+5)- (10+100)= 35
 Potasyum (5 mmol/L) Bikarbonat (10 mmol/L) Klor (100 mmol/L) (140+5)- (10+100)= 35.")
42
Anyon açığının artması mevcut metabolik asidozun, organik asit birikimine bağlı olduğunu gösterir

43
Anyon açığının normal olduğu (hiperkloremik) bir metabolik asidoz örneği
Sodyum (140 mmol/L) Potasyum (5 mmol/L) Bikarbonat (10 mmol/L) Klor (115 mmol/L) (140+5)-(10+115)= 20
 Potasyum (5 mmol/L) Bikarbonat (10 mmol/L) Klor (115 mmol/L) (140+5)-(10+115)= 20.")
44
Anyon açığının normal oluşu mevcut metabolik asidozun bikarbonat kaybına bağlı olduğunu gösterir

45
Organik asidemiler: Patogenez
İlgili CoA metabolitlerinin mitokondriyal birikiminin toksik etkileri + enerji eksikliği

46
Organik asidemiler: Klinik bulgular
Letarji Koma Trunkal hipotoni Ekstremite hipertonisitesi Miyoklonik sıçramalar Beslenme sorunları Beyin ödemi Dehidratasyon Anormal koku

47
Organik asidemiler: Laboratuvar bulguları ve tanı
Asidoz(anyon açığının artması) Hiperammmonemi Hiperlaktatemi Tandem-MS İdrarda organik asitler (GC-MS) Enzim ve DNA incelemeleri
 Hiperammmonemi. Hiperlaktatemi. Tandem-MS. İdrarda organik asitler (GC-MS) Enzim ve DNA incelemeleri.")
48
METABOLİK ASİDOZ Anyon açığı artışı Organik asidemi Ketozis Var Yok
Renal tübüler asidoz Anyon açığı artışı Yok (hiperkloremi) Gastroenteritler Var (normokloremi) Metilmalonik asidemi Organik asidemi * * Propionik asidemi * Ketotiolaz yetersizliği * İzovalerik asidemi (özel koku) Ketozis Var * Çoğul karboksilaz yetersizliği (deri döküntüleri) * Biotinidaz yetersizliği (deri döküntüleri, kellik) Yok * Akçaağaç şurubu kokulu idrar hastalığı (çemen kokusu) 3 - hidroksi - 3 - metilglütarik asidemi Çoğul açil CoA dehidrogenaz yetersizliği (Özel koku) Pirüvat karboksilaz yetersizliği ( hiperlaktatemi, hipoglisemi) Pirüvat dehidrogenaz yetersizliği ( hiperlaktatemi, hipoglisemi) MCAD ve LCAD yetersizlikleri (hiperammonemi, hipoglisemi)
 Gastroenteritler. Var (normokloremi) Metilmalonik asidemi. Organik asidemi. * * Propionik asidemi. * Ketotiolaz yetersizliği. * İzovalerik asidemi (özel koku) Ketozis. Var. * Çoğul karboksilaz yetersizliği. (deri döküntüleri) * Biotinidaz yetersizliği. (deri döküntüleri, kellik) Yok. * Akçaağaç şurubu kokulu idrar. hastalığı (çemen kokusu) 3. - hidroksi metilglütarik asidemi. Çoğul açil CoA dehidrogenaz yetersizliği (Özel koku) Pirüvat karboksilaz yetersizliği ( hiperlaktatemi, hipoglisemi) Pirüvat dehidrogenaz yetersizliği ( hiperlaktatemi, hipoglisemi) MCAD ve LCAD yetersizlikleri (hiperammonemi, hipoglisemi)")
49
Organik asidemiler: tedavi
Akut Toksinlerin uzaklaştırılması: Dializ, hemofiltrasyon, kan değişimi Bikarbonat Katabolizmanın önlenmesi Protein alımının önlenmesi Karnitin ( mg/kg) Vitaminler (Vit. B12, Vit. B1, Vit. B2, biotin) Kronik Proteinden kısılı diyet (varsa özel formülalar) Karnitin
 Vitaminler (Vit. B12, Vit. B1, Vit. B2, biotin) Kronik. Proteinden kısılı diyet (varsa özel formülalar) Karnitin.")
51
- ilerleyici ansefalopati, - beyin ödemi, letarji Tanı
Akçaağaç Şurubu Kokulu İdrar Hastalığı (Maple Syrup Urine Disease=MSUD) Klinik bulgular - ilerleyici ansefalopati, - beyin ödemi, letarji Tanı İdrar ve terde çemen kokusu, İdrarda DNPH testi pozitifliği (nonspesifik) Aminoasit analizi: valin, lösin, izolösin ve alloizolösin (diagnostik) artışı.
 Klinik bulgular. - ilerleyici ansefalopati, - beyin ödemi, letarji. Tanı. İdrar ve terde çemen kokusu, İdrarda DNPH testi pozitifliği (nonspesifik) Aminoasit analizi: valin, lösin, izolösin ve alloizolösin (diagnostik) artışı.")
52
Akçaağaç Şurubu Kokulu İdrar Hastalığı: Tedavi
Akut Anabolizmanın artırılması: Glükoz + insülin Detoksifkasyon (diyaliz, kan değişimi): Her zaman gerekli değil B1 (tiamin): 5-10 mg/kg/gün Kronik Diyet (lösin düzeyini izleyin, izolösin ve valin eksikliğine dikkat!) ± B1 vitamini Prognoz: ilk iki haftada tedavi edilmezse ölüm ve ağır sekel riski büyük!
: Her zaman gerekli değil. B1 (tiamin): 5-10 mg/kg/gün. Kronik. Diyet (lösin düzeyini izleyin, izolösin ve valin eksikliğine dikkat!) ± B1 vitamini. Prognoz: ilk iki haftada tedavi edilmezse ölüm ve ağır sekel riski büyük!")
53
Sıkça görülen bazı organik asidopatilerde klinik ve laboratuar bulguları
İzovalerik asidemi Ketoasidoz, dehidratasyon, nötropeni, trombositopeni, hiperammonemi, terli ayak kokusu Propionik asidemi Psikomotor gerilik, ketoasidoz, dehidratasyon, nötropeni, trombositopeni, hiperammonemi, hipoglisemi Metilmalonik asidemi Psikomotor gerilik, ketoasidoz, nötropeni, trombositopeni, hiperammonemi, hipoglisemi, B12 vitaminine cevap(+)
")
54
Biotinidaz yetersizliği
Biotin (kompleks) Biotinidaz Biotin (serbest) pirüvat karboksilaz asetil CoA karboksilaz propionil CoA karboksilaz beta-metilkrotonil CoA karboksilaz
 Biotinidaz. Biotin (serbest) pirüvat karboksilaz. asetil CoA karboksilaz. propionil CoA karboksilaz. beta-metilkrotonil CoA karboksilaz.")
55
Biotinidaz yetersizliği
Sıklık Dünyada 1:60,000 Türkiyede 1:10,000 Klinik ve laboratuvar bulguları Ağır metabolik asidoz Alopesi Seboreik deri döküntüleri İnatçı konvülsiyonlar Tedavi 5-10 mg aktif biyotin ömür boyu verilir.

56
(açil CoA dehidrogenazlar)
Yağ asidi oksidasyonu Yağ asiti (plazma) Asetil CoA Yağ asiti (mitokondri) Karnitin Karnitin enzimleri Beta-oksidasyon (açil CoA dehidrogenazlar) 131 ATP Keton cisimleri HMG CoA- liaz HMG CoA- sentaz 3-ketotiolaz (tioforaz) Krebs döngüsü
 Asetil CoA. Yağ asiti (mitokondri) Karnitin. Karnitin enzimleri. Beta-oksidasyon. (açil CoA dehidrogenazlar) 131 ATP. Keton cisimleri. HMG CoA- liaz. HMG CoA- sentaz. 3-ketotiolaz (tioforaz) Krebs döngüsü.")
57
Yağ asidi oksidasyonu

58
Yağ asidi oksidasyonu defektleri
Karnitin taşınma defekti Karnitin döngüsü defektleri Karnitin palmitoiltransferaz I (CPTI) yetersizliği Karnitin translokaz yetersizliği Karnitin palmitoiltransferaz II (CPTII) yetersizliği ß- oksidasyon defektleri Çok uzun zincirli açil-CoA dehidrogenez (VLCAD) Orta zincirli açil-CoA dehidrogenez (MCAD) (en sık görülen) Kısa zincirli açil-CoA dehidrogenez (SCAD) Uzun zincirli hidroksiaçil-CoA dehidrogenez (LCHAD) Orta zincirli hidroksiaçil-CoA Kısa zincirli hidroksiaçil-CoA Keton yapım defektleri 3-HMG CoA liaz 3-HMG CoA sentaz
 yetersizliği. Karnitin translokaz yetersizliği. Karnitin palmitoiltransferaz II. (CPTII) yetersizliği. ß- oksidasyon defektleri. Çok uzun zincirli açil-CoA. dehidrogenez (VLCAD) Orta zincirli açil-CoA. dehidrogenez (MCAD) (en sık görülen) Kısa zincirli açil-CoA. dehidrogenez (SCAD) Uzun zincirli hidroksiaçil-CoA. dehidrogenez (LCHAD) Orta zincirli hidroksiaçil-CoA. Kısa zincirli hidroksiaçil-CoA. Keton yapım defektleri. 3-HMG CoA liaz. 3-HMG CoA sentaz.")
59
Yağ asidi oksidasyonu defektleri: Klinik bulgular (Reye-benzeri sendrom)
Ölümcül hipoketotik hipoglisemik koma (özellikle uzayan açlık, enfeksiyon, ve ameliyat gibi katabolik durumlarda) Karaciğer yetersizliği İskelet kası miyopatisi, kardiyomiyopati
 Karaciğer yetersizliği. İskelet kası miyopatisi, kardiyomiyopati.")
60
Yağ asidi oksidasyonu defektleri: Laboratuar bulguları
Ketonlar: Düşük Amonyak: yüksek Metabolik asidoz: Belirgin değil Glükoz: düşük-normal Transaminazlar: yüksek Protrombin zamanı: uzun İdrarda dikarboksilik asitlerin varlığı (GC-MS) Açilkarnitin profili (Tandem-MS, Tanısal) Enzim incelemeleri (Fibroblastlar, lenfositler)
 Açilkarnitin profili (Tandem-MS, Tanısal) Enzim incelemeleri (Fibroblastlar, lenfositler)")
61
Yağ asidi oksidasyonu defektleri: Tedavi
Akut tedavi Glükoz enfüzyonu (7-10 mg/kg/dakika), lipid verilmemeli! Karnitin (100mg/kg): Toksik ara metabolitlerin uzaklaştırılmasını sağlar. Karnitin döngüsü defekt-lerinde ve LCHAD yetersizliğinde zararlı olabilir. Kronik tedavi Uzun süren açlıktan kaçınma (sık beslenme), katabolizmayı arttıran durumların dikkatle izlenmesi
, lipid verilmemeli! Karnitin (100mg/kg): Toksik ara metabolitlerin uzaklaştırılmasını sağlar. Karnitin döngüsü defekt-lerinde ve LCHAD yetersizliğinde zararlı olabilir. Kronik tedavi. Uzun süren açlıktan kaçınma (sık beslenme), katabolizmayı arttıran durumların dikkatle izlenmesi.")
62
Laktik asidemiler

63
PİRÜVAT DEHİDROGENAZ (E1, E2, E3, fosfataz) YETERSİZLİĞİ
Semptom ve bulgular: Ansefalomiyopati (+++, +), asidozis (+++, +), ketozis (-), anormal BT ve MR bulguları Laktat/Pirüvat oranı normal ya da düşük (<10:1) Glükoz yükleme sırasında semptomlar agrave olur.
, asidozis (+++, +), ketozis (-), anormal BT ve MR bulguları. Laktat/Pirüvat oranı normal ya da düşük (<10:1) Glükoz yükleme sırasında semptomlar agrave olur.")
64
PİRÜVAT KARBOKSİLAZ YETERSİZLİĞİ (izole ya da mültipl)
Semptom ve bulgular: Ansefalomiyopati (+++), asidozis (+++, +), hiperammonemi (+++, +), Anormal BT ve MR bulguları Laktat/Pirüvat oranı >30 ß-hidroksibütirat/ asetoasetat: Normal ya da düşük (<1.5) Postprandiyal hiperketozis
, asidozis (+++, +), hiperammonemi (+++, +), Anormal BT ve MR bulguları. Laktat/Pirüvat oranı >30. ß-hidroksibütirat/ asetoasetat: Normal ya da düşük (<1.5) Postprandiyal hiperketozis.")
65
ELEKTRON TRANSPORT ZİNCİRİ VE OKSİDATİF FOSFORİLİZASYON

66
ELEKTRON TRANSPORT ZİNCİRİ HASTALIKLARI
Semptom ve bulgular: Ansefalopati, asidozis, ketozis ( ±), hiperammonemi, miyopati, kardiyomiyopati, Anormal BT ve MR bulguları Laktat/Pirüvat oranı >30/1, (Kompleks I, III ve IV) ß-hidroksibütirat/ asetoasetat oranı yüksek (Kompleks I, III ve IV) Tokluk(glükoz yükleme) sırasında laktat ve ß-hidroksibütirat artar. Açlık sırasında ketozis azalır (Paradoksal ketozis)
, hiperammonemi, miyopati, kardiyomiyopati, Anormal BT ve MR bulguları. Laktat/Pirüvat oranı >30/1, (Kompleks I, III ve IV) ß-hidroksibütirat/ asetoasetat oranı yüksek (Kompleks I, III ve IV) Tokluk(glükoz yükleme) sırasında laktat ve ß-hidroksibütirat artar. Açlık sırasında ketozis azalır (Paradoksal ketozis)")
67
1. Mitokondriyel DNA defektleri (parçalanmış kas lifleri var)
MELAS: ansefalomiyelopati, laktik asidoz, felç atakları MERRF: miyoklonik epilepsi, ansefalomiyelopati Kearns-Sayre: oftalmopleji, ansefalomiyelopati Pearson: Makrositer anemi, ansefalomiyelopati HCM: Hipertrofik kardiyomiyopati, miyopati 2. Nükleer DNA defektleri (parçalanmış kas lifleri yok) Sitokrom oksidaz yetersizliği NADH-CoQ redüktaz yetersizliği
 Sitokrom oksidaz yetersizliği. NADH-CoQ redüktaz yetersizliği.")
Benzer bir sunumlar