Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
HASTALIKLARIN BİYOKİMYASI

2
KARBONHİDRAT METABOLİZMASI BOZUKLUKLARI

3
FURUKTOZ METABOLİZMASI ve HASTALIKLARI

4
HEREDİTER FRUKTOZ İNTOLERANSI
Herediter fruktoz intoleransı, OR geçen ve fruktoz-1- fosfat aldolaz enziminin eksik olduğu bir metabolik hastalıktır. Fruktoz alımından sonra hipoglisemi ve dokuda fruktoz-1-fosfat birikimi ile karakterizedir. Bu birikim glikojen yıkımı yapan glikojen fosforilazı inhibe eder. Bu nedenle çocuklarda fruktozla tetiklenen hipoglisemi oluşur. Çocuklar şeker almadıkları için dişlerinde çürüme olmaz.

5
Diğer önemli bulguları;
Gelişme geriliği Kusma Sarılık Hepatomegali Proteinüri Böbrekte Fankoni sendromu (Fosfatüri, bikarbonatüri, aa.üri, glukozüri) Tedavi edilmeyen vakalarda karaciğer yetmezliği sonucu ölüm görülür.
 Tedavi edilmeyen vakalarda karaciğer yetmezliği sonucu ölüm görülür.")
6
Bir diğer fruktoz metabolizma bozukluğu olan Esansiyel Fruktozüride ise Fruktokinaz eksikliği vardır. Klinik tablo olarak daha selim gidişlidir ve prognozu iyidir. İdrarda reduktan madde olarak çıkan fruktoz, yanlış DM teşhisi koydurabilir.

7
GALAKTOZ METABOLİZMASI ve HASTALIKLARI

8
GALAKTOZEMİ Otozomal Resesif geçişlidir. Galaktoz 1- P Üridil Transferaz enzimi eksikliği vardır. Belirtiler yeni doğan döneminde dikkati çeker. Süt (laktoz içerir) ile beslenmeyi takiben karaciğer yetersizliği (bilirubinemi, koagülasyon bozuklukları, hipoglisemi) bulguları ortaya çıkar.
 ile beslenmeyi takiben karaciğer yetersizliği (bilirubinemi, koagülasyon bozuklukları, hipoglisemi) bulguları ortaya çıkar.")
9
Galaktoz-1-fosfatın KC, beyin ve renal tübüllerde birikmesi hepatik parankimal hastalık, mental retardasyon ve renal fankoni sendromuna yol açar. Galaktoz, aldoz reduktaz enzimi ile Galaktitol’ün lenste birikmesi de katarakta neden olur. Tedavisiz genellikle ilk birkaç ay içerisinde, özellikle E.coli sepsisine bağlı ölüm görülür. Yine tedavisiz vakalarda iki ayda, katarakt ve hepatik siroz gelişir.

10
En sık sarılık yapan metabolik hastalıktır
En sık sarılık yapan metabolik hastalıktır. Galaktoz overlere toksik olup, erken menapoz (< 35) olumasına neden olur. Galakto kinaz eksikliğinde ise diğer bulgular olmayıp sadece jüvenil katarakt ile prezente olur.
 olumasına neden olur. Galakto kinaz eksikliğinde ise diğer bulgular olmayıp sadece jüvenil katarakt ile prezente olur.")
11
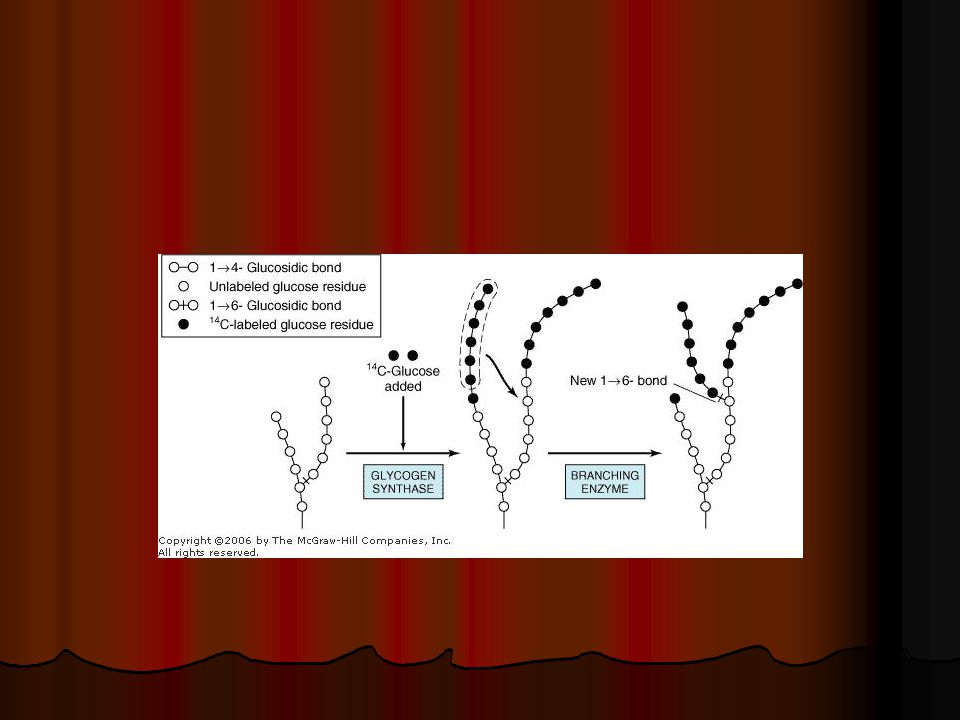
GLİKOJEN DEPO HASTALIKLARI
TIP I (Von Gierke hastalığı) Glukoz-6-fosfataz eksikliğidir. Sadece hepatik enzim yetersizliği vardır. İskelet veya kalp kası tutulumu yoktur. Bu hastalar yenidoğan döneminde semptomatik açlık hipoglisemisi, hafif hepatomegali ve hafif ketozis ile tanınırlar. Hepatomegali, laktat seviyesinin yüksekliği, hiperlipidemi (HiperTrigliseridemiye bağlı yanaklarda birikim sonucu doll face) hiperkolesterolemi , hiperürisemi ile beraber KC dokusunda enzim eksikliğinin gösterilmesi tanı koydurur.
 Glukoz-6-fosfataz eksikliğidir. Sadece hepatik enzim yetersizliği vardır. İskelet veya kalp kası tutulumu yoktur. Bu hastalar yenidoğan döneminde semptomatik açlık hipoglisemisi, hafif hepatomegali ve hafif ketozis ile tanınırlar. Hepatomegali, laktat seviyesinin yüksekliği, hiperlipidemi (HiperTrigliseridemiye bağlı yanaklarda birikim sonucu doll face) hiperkolesterolemi , hiperürisemi ile beraber KC dokusunda enzim eksikliğinin gösterilmesi tanı koydurur.")
12
TIP II (Pompe hastalığı)
Alfa1- 4 ve alfa1-6 glukozidaz eksikliği (Lizozomal alfa glukozidaz enzim eksikliği ) Lizozomal enzim kusuruna bağlı tek glikojen depo hastalığıdır. Klasik infantil formunda hastalık; Hızla gelişen konjestif kalp yetmezliği Kardiyomegali Kas hipotonisi Güçsüzlük ve makroglossi
 Lizozomal enzim kusuruna bağlı tek glikojen depo hastalığıdır. Klasik infantil formunda hastalık; Hızla gelişen konjestif kalp yetmezliği. Kardiyomegali. Kas hipotonisi. Güçsüzlük ve makroglossi.")
13
TIP III Amilo1-6 glukozidaz eksikliği (Debrancher enzim eksikliği, Forbes hastalığı, Cori hastalığı) KC, myokard ve iskelet kası tutulumu vardır. Klinik olarak Tip 1 den ayırt edilemez. Asidoz, hipoglisemi veya hiperlipidemi görülebilir. Mental gelişim normaldir

14
TIP IV Amilo1-4, 1- 6 transglukozidaz eksikliği (brancher enzim eksikliği, Anderson hastalığı); Birkaç aylıkken başlayan karaciğer sirozu ve erken dönemde karaciğer yetersizliğinden kaybedilir. Tüm dokular etkilenir. Mental gelişim normaldir.

16
TIP V Kas fosforilazı eksikliği (Mc Ardle hastalığı)
KC ve düz kaslar etkilenmez. Glikojen yıkılamadığı için egzersiz sırasında kramplar oluşur. Kas harabiyetine bağlı kas enzimleri (kreatin kinaz) yükselir. Egzersizler sonrasında, kan laktat düzeyinde yükselme görülmez.
 yükselir. Egzersizler sonrasında, kan laktat düzeyinde yükselme görülmez.")
17
ÜRE SİKLUS DEFEKTLERİ En sık OTC eksikliğidir. Temel semptomlar emmeme,laterji,taşipne ,apnedir. Glutamin birikimine bağlı beyin ödemi ,ensefalopati, beyin kanaması oluşur. Amonyağın solunum merkezini stimule etmesi sonucu respiratuar alkaloz sıktır. Tablo KC yetmezliğindeki hepatik ensafalopatiyi taklit eder. Kan amonyak yüksek, üre ise düşüktür.

18
Lipoprotein Metabolizma Bozuklukları Biyokimyası

19
Lipoprotein Metabolizma Bozuklukları
Hiperlipidemi Hipolipidemi

20
HİPERLİPİDEMİ: Toplum dağılımının en yüksek % 5-10’u
Trigliserit > 200 mg/dL Kolesterol > 240 mg/dL Çocuklarda trigliserit > 140 mg/dL kolesterol > 200 mg/dL

21
Lipit değerleri değişkendir
Yaş Genetik Beslenme Japonya’da kolesterol: 165 mg/dL Batı ülkelerinde: 210 mg/dL

22
Lipoproteinin Yapımında artış Uzaklaştırılmasında azalma

23
Lipoprotein Metabolizma Bozukluğu
Apolipoprotein Reseptör Enzim Kofaktör

24
Lipoprotein Metabolizma Bozukluğu
Birincil (genetik) İkincil (Diabetes Mellitus, hipotiroidizm, ilaçlar...) İkisi birlikte
 İkincil (Diabetes Mellitus, hipotiroidizm, ilaçlar...) İkisi birlikte.")
25
Fredrickson Sınıflandırması
Fenotip Bozukluk Lipit değişikliği Tip I Şilomikron Trigliserit Tip IIa LDL Kolesterol Tip IIb LDL - VLDL Tip III VLDL ve şilomikron kalıntısı Kolesterol ve trigliserit Tip IV VLDL Tip V Şilo - VLDL Trigliserit kolesterol

26
Dünya Sağlık Örgütü (WHO)
Tip Kolesterol LDL-K trigliserit Bozukluk I Yüksek Düşük-N Şilomikron IIa Yüksek-N Normal LDL IIb LDL-VLDL III kalıntıları ve IDL IV VLDL V Şilomikron ve VLDL

27
Lipoprotein fenotiplerinin etiyolojisi
Birincil neden İkincil neden I LPL, Apo CII eksikliği SLE IIa Ailesel Hiperkolesterolemi Hipotiroidizm IIb Ailesel kombine hiperlipidemi NS, DM, AN III Aillesel tip III hiperlipoproteinemi Hipotiroidizm, DM, Obesite IV Ailesel hipertrigliseridemi DM, KBY V Ailesel hipertrigliseridemi Apo CII eksikliği Alkol, diüretik, beta-bloker, OC

28
Ailesel Hiperkolesterolemi (Tip IIa)
LDL reseptöründe gen kusuru Plazma total kolesterol ve LDL-kolesterol artışı Total kolesterol heterozigotlarda 2-3 kat, homozigotlarda 3-6 kat artar Hiperlipidemi doğumla başlar Erken yaşta koroner kalp hastalığı görülür

29
Ailesel Hiperkolesterolemi (heterozigot)
1/500 Kolesterol > 300 mg/dL LDL-K > 250 mg/dL Trigliserit düzeyleri normal Koroner kalp hastalığı erkeklerde 45, kadınlarda 55 yaş öncesinde görülür

30
Ailesel Hiperkolesterolemi (homozigot)
1/ 10 yaşlarında KKH Tedavi edilmezsa 20’li yaşlarda MI Kolesterol > 600 mg/dL LDL-K > 550 mg/dL Planar ksantom

31
Lipoprotein Metabolizma Bozukluklarının Önemi
Hastaların tanısı Ailelerinin takibe alınması Lipoprotein metabolizmasının anlaşılması Atheroskleroz oluşumunun anlaşılması TUS sorularının yanıtlanması

32
Ailesel Defektif Apo B 100 Apo B 100 mutasyonu 1/500 - 700
Total kolesterol ve LDL-kolesterol yüksek Trigliserit normal Erken koroner kalp hastalığı riski yüksek Ailesel hiperkolesterolemiden daha hafif seyirli

33
Ailesel Kombine Hiperlipidemi
Sebebi bilinmiyor Orta derecede kolesterol ve/veya trigliserit artışı Hiperapobetalipoproteinemi Küçük yoğun LDL artmış Tip IIa, IV veya IIb Koroner kalp hastalığı riski yüksek Obesite, hiperürisemi, glukoz intoleransı ile birlikte olabilir Sendrom X’e benzerlik gösterir.

34
Sendrom X İnsülin direnci Küçük yoğun LDL’de artış
Trigliserit düzeylerinde artış HDL-K’de azalma

35
Tip III Hiperlipoproteinemi (ailesel disbetalipoproteinemi)
Apo E mutasyonu Kolesterolden zengin artık lipoproteinler birikir (beta VLDL) Kolesterol ve trigliserit mg/dl düzeylerindedir HDL-K’ün normal olması tipiktir LDL-K düşüktür Palmar ksantomlar patognomonik
 Kolesterol ve trigliserit mg/dl düzeylerindedir. HDL-K’ün normal olması tipiktir. LDL-K düşüktür. Palmar ksantomlar patognomonik.")
36
Tip III Hiperlipoproteinemi (ailesel disbetalipoproteinemi)
Erken yaşta koroner kalp ve periferik damar hastalığı görülür Obesite, alkol kullanımı, D.M. Ve hipotiroidizm tip III fenotipini arttırır

37
Lipoprotein Lipaz Eksikliği
LPL gen mutasyonu Bebek veya çocukta tekrarlayan karın ağrısı veya pankreatit ve hipertrigliseridemi Plazma trigliserit düzeyi > 1000 mg/dL (şilomikronemi sendromu) Döküntülü ksantomlar ve lipemia retinalis
 Döküntülü ksantomlar ve lipemia retinalis.")
38
Lipoprotein Lipaz Eksikliği
Plazma bir gece buzdolabında beklediğinde üstte krema gibi şilomikronlar, altında bulanıklık ise VLDL’dir Homozigot 1/ Yarı normal LPL 1/500

39
Apo C II Eksikliği LPL eksikliğine benzer bir tablodur
Otozomal resesif geçişlidir

40
Ailesel Hipertrigliseridemi
Apo B normal, trigliserit yapımı aşırı Trigliseritten zengin büyük VLDL HDL-kolesterol düzeyleri düşük Plazma kolesterolü referans aralıkta

41
Ailesel Hipoalfalipoproteinemi
HDL-kolesterolü düşüktür Erkeklerde < 30 , kadınlarda < 40 mg/dL Erken yaşta ve artmış koroner kalp hastalığı riski Nedeni bilinmemektedir

42
Apo AI mutasyonları HDL-K < 10 mg/dL
Koroner kalp hastalığı ile ilişkisi yok Ksantom Korneal opasite

43
Kolesterol Ester Transfer Protein Eksikliği
Azalmış CETP aktivitesisonucu homozigotlarda HDL-K > 100 mg/dL Koroner kalp hastalığında olası korunma ?

44
Lesitin Kolesterol Açil Transferaz Eksikliği
Korneal opasite Normokromik anemi Böbrek yetmezliği Hem periferde hem de lipoprotein partiküllerinde kolesterol birikimi olur Kolesterol seviyeleri değişkendir HDL-kolesterol düzeyleri çok düşüktür

45
Balık Gözü Hastalığı LCAT eksikliğinin hafif formudur
Anemi, böbrek yetmezliği, erken koroner kalp hastalığı riski yoktur

46
Tangier Hastalığı HDL ve LDL kolesterol düzeyleri düşüktür
Portakal rengi tonsiller Korneal opasite Hepatosplenomegali Periferik nöropati Aşırı miktarda kolesterol esteri RES makrofajlarında birikir ABC-1 mutasyonu

47
Aielesel Hipobetalipoproteinemi
Heterozigotlarda kolesterol ve LDL-K düzeyleri normalin yarısı kadar, homozigotlarda < 50 mg/dL Heterozigot 1/500 HDL-K normal veya hafif artmış Yağ malabsorbsiyon bulguları (yağda eriyen vitaminlerin emilememesi sonucu)
")
48
Abetalipoproteinemi Apo B-içeren lipoproteinler yok 1 /

49
İkincil Hiperlipidemi Nedenleri
Hormonal etkiler Metabolik bozukluklar Böbrek hastalıkları Tıkanma tipi karaciğer hastalıkları Toksinler iyatrojenik

50
Hormonal Etkiler Gebelik Ekzojen seks hormonları Hipotiroidizm

51
Metabolik Bozukluklar
Diabetes Mellitus Gut Obesite Progresif parsiyel lipodistrofi Depolama bozuklukları

52
Böbrek Hastalıkları Nefrotik Sendrom
Kronik Böbrek Yetmezliği, diyaliz veya transplantasyon sonrası

53
Toksinler Alkol Dioksin veya klorlu hidrokarbonlar

54
İyatrojenik Antihipertansifler İmmunosüpresanlar Diğer ilaçlar

55
Endokrin/Metabolik ikincil hiperlipidemi nedenleri
Hastalık lipoprotein Mekanizma (?) Diabetes M. VLDL, şilo yapım, yıkım Hipotiroidizm LDL temizlenme Östrojen VLDL yapım Glukokortikoid VLDL, LDL VLDL yapımı LDL dönüşümü Hipopituitarizm Akromegali A. nervoza safra atılımı Lipodistrofi
 Diabetes M. VLDL, şilo. yapım, yıkım. Hipotiroidizm. LDL. temizlenme. Östrojen. VLDL. yapım. Glukokortikoid. VLDL, LDL. VLDL yapımı LDL dönüşümü. Hipopituitarizm. Akromegali. A. nervoza. safra atılımı. Lipodistrofi.")
56
Diabetes Mellitus Hipertrigliseridemi 1/3 vakada HDL-K düşük
İnsülin eksikliği LPL aktivitesinde azalma Lipolizde artma Karaciğerde VLDL yapımı artar LDL de artabilir

57
Endokrin dışı ikincil hiperlipidemi nedenleri
Hastalık lipoprotein Mekanizma (?) Alkol VLDL, şilo Yapım Nefrotik Send. VLDL, LDL VLDL yapımı Üremi VLDL Temizlenme Safra tıkanıklığı LP-X Safra K ve FL dolaşıma geçmesi Hepatit LCAT SLE şilo LPL aktiv. Monoklonal gamopati VLDL, IDL, LDL Yıkım
 Alkol. VLDL, şilo. Yapım. Nefrotik Send. VLDL, LDL. VLDL yapımı. Üremi. VLDL. Temizlenme. Safra tıkanıklığı. LP-X. Safra K ve FL dolaşıma geçmesi. Hepatit. LCAT. SLE. şilo. LPL aktiv. Monoklonal gamopati. VLDL, IDL, LDL. Yıkım.")
58
PORFİRYALAR

59
Akut intermittent porfirya
Otozomal dominant geçer. Kadınlarda sıktır. Porfobilinojen deaminaz (Üroporfirinojen sentaz) eksiktir. İdarda aminolevulinik asit ve porfobilinojen atılımı artar. İdrar havaya ve ışığa maruz kalınca koyulaşır. Genellikle asemptomatiktir ancak, ALA sentazı aktive eden enzim induktörleri hastalığı semptomatik hale getirir. Östrojen, sülfonamid ve barbitürat alınımıyla semptomatik hale geçebilir.
 eksiktir. İdarda aminolevulinik asit ve porfobilinojen atılımı artar. İdrar havaya ve ışığa maruz kalınca koyulaşır. Genellikle asemptomatiktir ancak, ALA sentazı aktive eden enzim induktörleri hastalığı semptomatik hale getirir. Östrojen, sülfonamid ve barbitürat alınımıyla semptomatik hale geçebilir.")
60
Porfirya kutanea tarda
UPG dekarboksilaz eksiktir. En sık görülen porfiryadır. Otozomal dominant geçer. Hexochlorobenzen ve polikloro hidrokarbonlar edinsel porfirya kutunea tarda’ya sebep olabilirler. İdrarda üroporfirin atılım artar.

61
YAĞ ASİDİ METABOLİZMA HASTALIKLARI

62
YAĞ ASİDİ MİTOKONDRİAL OKSİDASYON DEFEKTİ
Klinik bulgular kusma, hızla komaya ilerleyen letarji ve konvülziyonlardır. Aynı zamanda kas zayıflığı ve kardiyomiyopati vardır. Laboratuar bulguları: Nonketotik hipoglisemi, yüksek plazma NH3, ALT, AST ve uzun PT, PTT.

63
Yağ asitlerinin mikrozomal ve oksidasyonundan kaynaklanan idrarda orta zincirli dikarboksilik asit düzeylerinde artış olur. Sekonder karnitin eksikliği meydana gelir. Tedavi: Akut atakta acilen % 10 dekstroz İV. verilerek lipoliz baskılanmaya çalışılır. Kronik tedavi hastaların asla saatten fazla aç kalmamaları ile sağlanır.

64
REFSUM HASTALIĞI Alfa oksidaz eksikliğidir. Refsum hastalığında dallızincirli yağ asidi olan fitanik asid birikir. Fitanik Asid insanda bulunmaz. İnek sütünden alınır. Refsum Semptomları : Retinitis pigmentoza İktiyozis Gece körlüğü Periferal nöropati (8. kranial sinir tutumuna bağlı sağırlık oluşur) Ataksi
 Ataksi.")
65
ZELLWEGER SENDROMU Tipik yüz görünümü (yüksek alın, çekik gözler, hipoplastik kaş kavsi ve epikantal kıvrımlar) Hipotoni ve”mongoloid” görünüm nedeniyle Down sendromu ile karışabilir. Neonatal konvülziyonlar, psikomotor retardasyon Göz bulguları (katarakt, glokom, kornea bulanıklığı, Brushfield lekeleri, pigmenter retinopati, optik sinir displazisi). Mikronodüler siroz, hepatomegali, kolestaz, renal kistler bulunur. Zellweger’li bebekler nadiren birkaç aydan fazla yaşar.
 Hipotoni ve mongoloid görünüm nedeniyle Down sendromu ile karışabilir. Neonatal konvülziyonlar, psikomotor retardasyon Göz bulguları (katarakt, glokom, kornea bulanıklığı, Brushfield lekeleri, pigmenter retinopati, optik sinir displazisi). Mikronodüler siroz, hepatomegali, kolestaz, renal kistler bulunur. Zellweger’li bebekler nadiren birkaç aydan fazla yaşar.")
66
ADRENOLOKODİSTROFİ X’e bağlı geçiş gösteren adrenolökodistrofi, doymuş uzun zincirli yağ asidlerinin birikimi, adrenal korteks ve sinir sistemi beyaz cevherinin progressif fonksiyon bozukluğu ile karakterizedir. Esas bozukluk çok uzun zincirli doymuş yağ asidlerinirı (C24- C26) birikmesidir. Bu yağ asidlerinin peroksizomlardaki yıkım kapasitesi azalmıştır. Peroksismal lignoceroyl CoA ligazın fonksiyonu bozuktur.
 birikmesidir. Bu yağ asidlerinin peroksizomlardaki yıkım kapasitesi azalmıştır. Peroksismal lignoceroyl CoA ligazın fonksiyonu bozuktur.")
67
Klinik: İlk 3- 4 yaşlarda fenotipik özellikler normaldir, Daha sonra hastalığa özgü bulgular çıkar. ALD’de ilk semptomlar 4- 8 yaşları arasında ortaya çıkar. İlk ortaya çıkan semptom hiperaktivitedir. Daha sonra başarılı bir öğrenci iken okul performansının bozulduğu görülür. İşitme fonksiyonu bozulur. Ataksi, el becerilerinde bozulma, konvülziyon ortaya çıkar. ALD’de serebral bulgular hızla ilerler, spastisite, paralizi, görme ve işitme bozukluğu, konuşma ve yutkunmanın bozulması belirginleşir. Bulguların ağırlaşması ortalama iki yıl alır. Addison ve hiperpigmentasyon tabloya eklenir.

68
GENETİK HASTALIKLARIN BİYOKİMYASI VE TESTLERİ

69
Genler: Kalıtım birimleri Protein sentezini kodlayan DNA dizilimleri

70
Genetik hastalıklar: Genlerdeki mutasyonların sonucu olarak ortaya çıkan patolojik durumlar

71
Mutasyon somatik hücrelerde etkili olursa edinsel hastalık ortaya çıkar
Mutasyon germ kökenli hücrelerde etkili olursa otozomal (dominant veya resesif) veya cinsiyete bağlı (tipik olarak X kromozomu ile ilgili) kalıtılan kalıtsal hastalık ortaya çıkar
 veya cinsiyete bağlı (tipik olarak X kromozomu ile ilgili) kalıtılan kalıtsal hastalık ortaya çıkar.")
72
Genetik hastalıklar, Üç grup altında incelenirler: -Kromozomal bozukluklar -Monogenik bozukluklar -Poligenik (multifaktöryel) bozukluklar
 bozukluklar.")
73
Kromozomal bozukluklar:
Kromozomların sayı veya dizilimindeki değişiklikler nedeniyle eksik veya fazla genetik materyal oluşması Çoğu (hepsi değil) edinsel hastalıklara neden olur ve kalıtılmaz Anöploidi: Bir veya daha fazla kromozomun kaybı veya fazlalığıdır. Trizomi, bir kromozomun 2 yerine 3 kopyasının bulunması. Monozomi, bir kromozomun çift yerine tek kopya olması…
 edinsel hastalıklara neden olur ve kalıtılmaz. Anöploidi: Bir veya daha fazla kromozomun kaybı veya fazlalığıdır. Trizomi, bir kromozomun 2 yerine 3 kopyasının bulunması. Monozomi, bir kromozomun çift yerine tek kopya olması…")
74
Monogenik bozukluklar: Bir genin veya gen lokusunun hatası
Kalıtsal hastalık ile sonuçlanması en olası bozukluk Mutasyonların çoğunluğu Tek nükleotid eklenmesi (nokta mutasyonu) Daha fazla nükleotidin eklenmesi veya çıkarılması (çerçeve kayması mutasyonu)
 Daha fazla nükleotidin eklenmesi veya çıkarılması (çerçeve kayması mutasyonu)")
75
Poligenik (multifaktöryel) bozukluklar:
Fiziksel ve kimyasal faktörlerle bozulmuş çok sayıda genin etkileşimi sonucu oluşan bozukluklar Ailelerden görülür Kalıtım modeli öngörülemez ve tekrarlama riski tipik Mendelyen genetik bozukluklara bağlı olarak %25-50’den azdır Alzheimer hastalığı, koroner arter hastalığı, belirli kanserler, tip II diabetes mellitus, hipertansiyon gibi kompleks hastalıkların poligenik (multifaktöryel) bozukluk olduğu düşünülmektedir
 bozukluk olduğu düşünülmektedir.")
76
Genom, genotip, fenotip:
İnsan genomu: Her çekirdekli hücrede 22 çift otozom ve 1 çift seks kromozomu (diploid sayı-diploid hücre) Kromozomların içindeki bireysel gen dizilimleri genotipi oluşturur Bir genin değişik şekilleri alel olarak adlandırılır Çiftteki aleller aynı dizilime sahipse homozigot, farklı dizilime sahipse heterozigot alellerdir
 Kromozomların içindeki bireysel gen dizilimleri genotipi oluşturur. Bir genin değişik şekilleri alel olarak adlandırılır. Çiftteki aleller aynı dizilime sahipse homozigot, farklı dizilime sahipse heterozigot alellerdir.")
77
Sperm ve ovum, bir sonraki döle aktarmak için gereken genetik materyalin yarısını taşırlar (haploid sayı-haploid hücreler) Sperm ve ovum’un birleşmesi, erken embriyonik hücrelerde görülen 23 çift kromozomu oluşturur

78
Genotipin ekspresyonu ile fenotip ortaya çıkar
Hastalık fenotipleri göz önüne alındığında; Baskınlık (dominans), kromozomun belirli lokusunda heterozigot alellerin varlığı sonucunda görülen fenotipik özelliktir. Etkilenmiş bireyler otozomal dominant hastalık genine ve normal gene sahiptir
, kromozomun belirli lokusunda heterozigot alellerin varlığı sonucunda görülen fenotipik özelliktir. Etkilenmiş bireyler otozomal dominant hastalık genine ve normal gene sahiptir.")
79
Çekiniklik (resesif), kromozomun belirli lokusunda homozigot alellerin varlığı sonucunda görülen fenotipik özelliktir Aleller otozomlar üzerinde yerleşmişse fenotipler otozomal dominant özellikler veya otozomal resesif özellikler olarak adlandırılır; eğer X kromozomu üzerinde yerleşmişse X’e bağlı dominant özellikler veya X’e bağlı resesif özellikler olarak adlandırılır

80
Geleneksel kalıtım modelleri: Otozomal dominant kalıtım modeli:
1. Özellikler dikey olarak (soyağacında üstten alta) ebeveynden çocuğa aktarılır 2. Kadın ve erkekler kalıtım açısından eşit şansa sahiptir 3. Özelliğe sahip olan bireyin bunu çocuklarına geçirme olasılığı %50’dir 4. Özelliğe sahip olmayan bireylerin çocukları da özelliğe sahip olmaz 5. Özelliğe sahip bireylerin aileleri de özelliğe sahiptir Bazen, otozomal dominant bozukluğu olan bir üyeye sahip ailenin soyağacı listelenen 5 özelliği izlemez
 ebeveynden çocuğa aktarılır. 2. Kadın ve erkekler kalıtım açısından eşit şansa sahiptir. 3. Özelliğe sahip olan bireyin bunu çocuklarına geçirme olasılığı %50’dir. 4. Özelliğe sahip olmayan bireylerin çocukları da özelliğe sahip olmaz. 5. Özelliğe sahip bireylerin aileleri de özelliğe sahiptir. Bazen, otozomal dominant bozukluğu olan bir üyeye sahip ailenin soyağacı listelenen 5 özelliği izlemez.")
81
Otozomal resesif kalıtım modeli:
1. Soyağacında özellik yatay olarak görülür 2. Homozigot alele sahip etkilenmiş bireylerin etkilenmemiş heterozigot ebeveynleri vardır 3. Heterozigot ebeveynlerin homozigot, etkilenmiş çocuk sahibi olma olasılığı %25’dir Kistik fibrozis, orak hücreli anemi, PKU, Tay Sach’s hastalığı, otozomal resesif kalıtım modeli gösteren hastalıklardır. Enzim bozukluklarının (doğuştan metabolik hastalıklar) çoğu otozomal resesiftir
 çoğu otozomal resesiftir.")
82
X’e bağlı kalıtım modeli:
1. Taşıyıcı kadın %50 olasılıkla normal X kromozomunu kız veya erkek çocuğa aktarır 2. Etkilenmiş erkeklerin tüm erkek çocukları sağlam, tüm kız çocukları ise taşıyıcıdır 3. Taşıyıcı kadın %25 olasılıkla etkilenmiş erkek çocuk, etkilenmemiş erkek çocuk, taşıyıcı olmayan kız çocuk ve taşıyıcı kız çocuk sahibi olur Hemofili, Duchene müsküler distrofisi (DMD) ve frajil X sendromu X’e bağlı bozukluk örnekleridirler
 ve frajil X sendromu X’e bağlı bozukluk örnekleridirler.")
83
Geleneksel olmayan kalıtım modelleri:
Uniparental dizomi: Bir bireyin her iki kromozom kopyasının bir ebeveyninden gelmesi durumunda… İmprinting: Çocukta genin anneden veya babadan kalıtılması ile belirlenen tercihli ekspresyon Mozaizm: İki veya daha fazla hücre tipi varlığında birinin normal, diğerinin anormal genetik yapıya sahip olması Antisipasyon: Üçlü tekrarların genişlemesine bağlı olarak hastalık başlangıcının beklenenden daha erken olması

84
Mitokondriyal kalıtım modeli:
mtDNA tümüyle ovum’un sitoplazmasındadır ve kalıtım şekli dikeydir Yaklaşık 50 zararlı mtDNA bozukluğu erkek ve kzı çocukları etkiler

85
Özel kalıtsal hastalıklar:
-Neoplastik hastalıklar -Kardiyovasküler hastalıklar Faktör V mutasyonu -Nöromüsküler hastalıklar -Nörolojik hastalıklar Huntington hastalığı Frajil X sendromu Ailesel erken başlangıçlı Alzheimer hastalığı -Doğuştan metabolik hastalıklar Lizozomal depo hastalıkları -Diğer sık rastlanan kalıtsal bozukluklar Herediter hemokromatoz Kistik fibrozis

86
Kalıtsal hastalıkların klinik özellikleri:
Bir çoğu klinikte anormallik yaratmaz Genetik danışmanlık, prenatal tanı ve gebeliğin sonlandırılması açısından önemlidir Uygulanacak tedavi, geri dönüşümsüz klinik sonuçları veya ölümü önler

87
Kalıtsal hastalıkların klinik laboratuvarda değerlendirilmesi:
-Kromozom analizleri -DNA analizleri: Gen dizilimleri kalıtsal hastalıklardaki mutasyonların belirlenmesi -Enzim aktivitesi yokluğu veya azlığının belirlenmesi -Öncül proteinlerde konsantrasyon artışının belirlenmesi -Birikmiş metabolitin değerlendirilmesi -Enzim-substrat reaksiyon ürününün eksikliğinin belirlenmesi

88
Prenatal tarama testleri:
Yüksek risk gruplarında; -Daha önce hasta bir çocuk doğurmuş olan kadınlar -Taşıyıcılığın yüksek olduğu etnik gruplar Sadece bazı hastalıklar için: Amniosentez veya korionik villus biopsisiyle alınan fibroblastlar kültüre ekilir, üreyen hücrelerde metabolik defekt gösterilir.

89
Yenidoğanda tarama testleri:
Doğumdan sonraki 6-9. günlerde topuktan süzgeç kağıdına alınan kan, 4-6. haftalarda alınan idrarda -Fenilketonüri -Hipotiroidizm -Galaktozemi -Kistik fibrozis için… Pozitif test yenilenmeli, nicel testler yapılmalı

90
Bebeklik veya erken çocuklukta tarama testleri:
Kusma Gelişme geriliği Hepatosplenomegali Uzun süren sarılık Mental retardasyon, epileptik nöbet Çamaşır veya çarşaflarda özel koku veya renk Hipoglisemi Metabolik asidoz Tedaviye dirençli raşitizm Böbrek taşları varlığında…

91
Doğuştan metabolizma bozukluklarının tanısı için dolaylı tarama testleri:
-Üre döngüsü bozukluklarında plazma amonyak düzeyi ölçümü -Amino asit metabolizması bozukluklarında plazma ve idrarda amino asit kromatografisi Dallı zincirli amino asit metabolizma bozukluğunda idrarda organik asit aranması

92
Karbohidrat metabolizması bozuklukları ve laboratuvar:
-Glikojen depo hastalıkları (tip I-IX glikojenoz) -Galaktozemi -Fruktoz metabolizması bozuklukları Kalıtsal fruktoz intoleransı Esansiyel fruktozüri Genellikle hipoglisemi ve kas güçsüzlüğü ile karakterizedirler
 -Galaktozemi. -Fruktoz metabolizması bozuklukları. Kalıtsal fruktoz intoleransı. Esansiyel fruktozüri. Genellikle hipoglisemi ve kas güçsüzlüğü ile karakterizedirler.")
93
Amino asit metabolizması bozuklukları ve laboratuvar:
Fenilketonüri: Fenilalanin hidroksilaz eksikliği veya yokluğu Guthrie tarama testi: Bacillus subtilis adlı organizmanın emdirildiği disk şeklindeki filtre kağıdına topuk kanının uygulanmasıyla mikroorganizmanın üremesi testin pozitif olduğu anlamına gelir. Test, kan fenilalanin düzeyi ölçümüyle doğrulanmalıdır

94
Alkaptonüri: Homogentisik asit oksidaz eksikliği
İdrarla homogentisik asit atılır; idrar bekletilince koyulaşır

95
Ardıç ağacı şurubu idrar hastalığı (Maple syrup Urine Disease, MSUD): Dallı zincirli amino asitlerin metabolizması sırasında oluşan ketoasitleri dekarboksile eden enzim eksikliği İdrarla atılan ketoasitler, idrarın karakteristik ardıç ağacı şurubuna benzeyen kokusundan sorumludurlar Laboratuvar araştırmaları sırasında hastalarda metabolik asidoz ve hipoglisemiye rastlanır Tanı, plazmada aşırı miktarda yükselmiş dallı zincirli amino asit düzeylerinin saptanmasıyla konur

96
Organik asidemiler ve laboratuvar:
Amino asit, karbohidrat ve lipid metabolizması sırasında sentezlenen düşük molekül ağırlıklı karboksilik asitlerin birikmesi Hipoglisemi, ketozis, laktik asidozis ve hiperammonemi görülür Tanı, idrarda özgün organik asit düzeyinin saptanması ve enzim aktivitelerinin belirlenmesiyle konur

97
Hiperammonemiler ve laboratuvar:
Üre döngüsünde yer alan enzimlerden birinin eksikliğine bağlı Kan amonyak düzeyi yüksekliği

98
Reye sendromu ve laboratuvar:
15 yaşın altındaki çocuklarda, genellikle bir ust solunum yolu enfeksiyonunu takiben kusma, konvülsiyon ve koma Serum transferazları ve amonyak düzeyi yüksekliği, metabolik asidoz, uzamış protrombin zamanı ve hipoglisemi başlıca laboratuvar bulgularıdır

99
Kistik fibrozis ve laboratuvar:
Kistik fibrozis membran iletkenlik düzenleyicisindeki (CFTR) kusur nedeniyle Salgıların su içeriği azalmıştır ve normalden daha koyu kıvamda bulunurlar Tanı, karakteristik klinik bulgularla ve ter klor konsantrasyonunun 60 mmol/L’nin üstünde olmasıyla konur
 kusur nedeniyle. Salgıların su içeriği azalmıştır ve normalden daha koyu kıvamda bulunurlar. Tanı, karakteristik klinik bulgularla ve ter klor konsantrasyonunun 60 mmol/L’nin üstünde olmasıyla konur.")
100
Lizozomal depo hastalıkları ve laboratuvar:
Lizozomlardaki hidrolitik enzimlerin bir veya daha fazlası tarafından yıkılabilecek lizozomal metabolitlerin birikmesi sonucu… -Niemann-Pick hastalığı (sfingomiyelin lipidoz): Sfingomyelinaz eksikliği -Gaucher hastalığı (glukozilseramid lipidoz): β-glukozidaz eksikliği -Krabbe hastalığı (galaktozilseramid lipidoz): Galaktozilseramid β-glukozidaz eksikliği -Fabry hastalığı: α-galaktozidaz eksikliği -Tay Sach’s hastalığı (GM2-gangliozidoz): β-N-asetil Glukozaminidaz A (NAG-A) eksikliği
: Sfingomyelinaz eksikliği. -Gaucher hastalığı (glukozilseramid lipidoz): β-glukozidaz eksikliği. -Krabbe hastalığı (galaktozilseramid lipidoz): Galaktozilseramid β-glukozidaz eksikliği. -Fabry hastalığı: α-galaktozidaz eksikliği. -Tay Sach’s hastalığı (GM2-gangliozidoz): β-N-asetil Glukozaminidaz A (NAG-A) eksikliği.")
101
İlaç duyarlılıkları ve laboratuvar:
-Suksametonyum duyarlılığı: Psödokolinesteraz aktivitesi düşüklüğü… -Bazı sıtma ilaçları ve K vitamini analoglarına bağlı hemolitik kriz: Glukoz-6-fosfatdehidrogenaz aktivitesi eksikliği

Benzer bir sunumlar















>")



