Sunuyu indir
Sunum yükleniyor. Lütfen bekleyiniz

1
İLAÇLARDA RUHSATLANDIRMA SÜRECİ Farmakoloji Ab.D. Öğ. Üyesi
Dr. Faruk ERDEN Kocaeli Üniversitesi Tıp Fakültesi Farmakoloji Ab.D. Öğ. Üyesi

2
Sunum İçeriği Etiket ve KÜB kavramları Ruhsatlandırma Nedir?
Ruhsatlandırmanın Genel Tanımı ve Konumu Dünyada Belli Başlı Kurumlar Ülkemizde Süreç Nasıl İlerlemektedir? Yeni Ürün Ruhsat Başvuru Dokümanları Türkiye’de İthal Ürün Lansman Süreci Türkiye’de Ruhsatın Tarihsel Gelişimi Yenilikçi İlaç Lansman Sürecindeki Kritik Basamak GMP Etiket ve KÜB kavramları

4
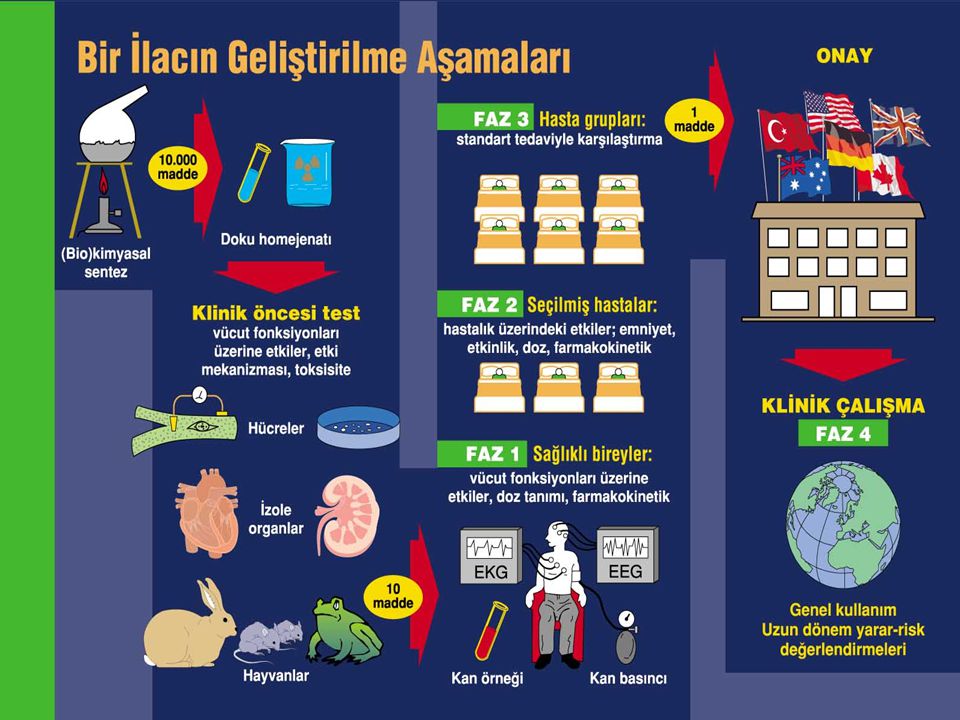
Ruhsatlandırma Nedir? Bir ilaç pazara sunulmadan önce, tüm dünyada, farmasötik endüstrisini düzenleyen farklı hükümet kurumlarından onay almalıdır. En etkili ruhsatlandırma kurumlarından biri Amerika Birleşik Devletleri Gıda ve İlaç İdaresi’dir. (Food and Drug Administration, FDA).
.")
5
FDA ABD’de ilaçların güvenliliğini ve etkililiğini düzenleyen geniş bir yetki alanına sahiptir. 10.000’den fazla çalışanı vardır. İlaçların üretimini, ithalatını, sevkiyatını, depolanmasını ve yıllık 1 trilyon doların üzerindeki satışını izler. Yakın tarihte Çin'de bir ofis açmıştır.

6
EMA ve PMDA Avrupa İlaç Kurumu (European Medicines Agency, EMA), Avrupa Birliği’ndeki 27 ülkede ilaçları denetim altına alan bir Avrupa Birliği kurumudur. EMA’nın varlığı Avrupa’da ilaçlar için tek bir ruhsatlandırma çerçevesi oluştururken, hem daha büyük bilimsel titizlik hem de düzgün bir karar verme süreci sağlar. PMDA, Japonya daki ruhsat otoritesidir.
, Avrupa Birliği’ndeki 27 ülkede ilaçları denetim altına alan bir Avrupa Birliği kurumudur. EMA’nın varlığı Avrupa’da ilaçlar için tek bir ruhsatlandırma çerçevesi oluştururken, hem daha büyük bilimsel titizlik hem de düzgün. bir karar verme süreci sağlar. PMDA, Japonya daki ruhsat otoritesidir.")
7
FDA FDA, iki yönlü bir role sahiptir. İlk olarak ve öncelikle, beşeri ilaçların güvenliliğini, etkililiğini ve emniyetini güvence altına alarak halk sağlığını korumaktan sorumludur. Ek olarak, ilaçları daha etkili, daha güvenli ve daha ekonomik hale getiren yenilikleri hızlandırmaya destek olmayı amaçlar. Dolayısıyla, bir yanda güvenliliğin sağlanması ile öte yanda yeniliklerin geliştirilmesi arasında dinamik bir gerilim söz konusudur.

8
ICH The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Avrupa, ABD ve Japonya’daki ruhsatlandırma otoritelerinin ve konuyla ilgili ilaç uzmanlarının bir araya gelerek konunun bilimsel ve teknik yönlerinin tartışılması projesidir.

9
ICH ve CTD 1990’lı yılların ortalarında ICH süreci sonuçlandırılarak kalite, güvenilirlik, etkinlik ve çok yönlülük alanlarında birçok kılavuz oluşturulmuştur. 2000 yılında ICH grubu tarafından Ortak Teknik Doküman (Common Technical Document - CTD) fikri ortaya çıkmıştır. CTD, zaman ve kaynak tasarrufunun sağlanması ile ruhsatlandırma incelemelerinin ve iletişimin kolaylaştırılmasını amaçlamaktadır. ICH teknik bir kılavuzdur, CTD ise bir format kılavuzudur.
 fikri ortaya çıkmıştır. CTD, zaman ve kaynak tasarrufunun sağlanması ile ruhsatlandırma incelemelerinin ve iletişimin kolaylaştırılmasını amaçlamaktadır. ICH teknik bir kılavuzdur, CTD ise bir format kılavuzudur.")
10
Ruhsatlandırma Derken…
ESAS SORUMLULUKLARI Keşif Ürün Gelişimi Ürün Ruhsatlandırılması Yeni ürün ruhsatlandırılması Ruhsat otoritesi ile iletişim Yönetmeliklerin takibi In vitro & deneysel çalışmalar İnsan çalışmaları/klinik çalışmalar Ürün formülasyonu Kalite & Güvenlilik Üretim Ambalajlama Otoritelerle iletişim Tüketiciye ürünün ulaşması Ruhsatlı ürünlerin yaşam döngüsünü sağlamak Onaylanmış bilgilerle ürünleri piyasaya sunmak Ürünle ilgili gelişmelerin takibi ve bildirimi RUHSATLANDIRMA Şirket kurallarına uyarak tüm süreçleri yürütmek

11
TÜRKİYE’DE RUHSATLANDIRMA
Ülkemizde ilaçların ruhsatlandırma işlemleri, Avrupa Birliği mevzuatına uyum çalışmaları çerçevesinde hazırlanan ve tarihinde yürürlüğe giren Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği hükümlerine göre yapılmaktadır. Yönetmelikte; ruhsat başvurusunda bulunabilecek ilgililer, başvurularda sunulması gereken dokümanlar, ruhsat başvurularının değerlendirilme, kabul ve red kriterleri, ruhsatın geçerlilik süresi, ruhsat sahibinin sorumlulukları, ruhsatın iptali, ruhsat sahibi değişikliği gibi konular yer almaktadır.

12
TÜRKİYE’DE RUHSATLANDIRMA
Sağlık Bakanlığı tarafından ruhsatlandırılmayan hiçbir beşeri tıbbi ürün pazara sunulamaz. Ayrıca, Türkiye sınırları dahilinde yerleşik bulunan gerçek veya tüzel kişiler, bir ürünü pazara sunmak amacıyla ruhsat alabilmek için gereken bilgi ve belgeleri, her bir farmasötik form için hazırlayarak Bakanlığa sunmaları gerekmektedir.

13
TÜRKİYE’DE RUHSATLANDIRMA
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinde Değişiklik Yapılmasına Dair Yönetmelik ise, tarih ve sayılı Resmi Gazete’de yayımlanarak yürürlüğe girmiş olup, 210 günlük ruhsatlandırma süresinin kısaltılmasına ilişkin bazı istisnalar getirilmiş, yenilenmiş olan AB direktifine uyum amacıyla bazı güncellemeler yapılmıştır.

14
TÜRKİYE’DE RUHSATLANDIRMA
Ülkemizde ilaçların ruhsatlandırılması için yapılacak işlemler ilacın referans veya eşdeğer olmasına göre farklılık göstermektedir.

15
Ruhsat Komisyonları İlaçların ruhsatlandırılması ve ruhsatlandırma sonrası işlemlerinde bilimsel danışmanlık yapmak üzere, uzmanlardan oluşan danışmanlık kurul listesinin teşkil edilmesi ve bu kuruldan belirlenecek komisyonların çalışma esasları, görev ve sorumlulukları, “Beşeri Tıbbi Ürünler Bilimsel Danışmanlık Kurulu ve Komisyonlarının Kuruluş ve Görevleri Hakkında Yönetmelik“ ile belirlenmiştir.

16
Paydaşlar Kimlerdir? Ruhsatlandırma Üretim Yerleri Komisyon Üyeleri
Pazar Erişimi Klinik Yönetim Ruhsatlandırma Medikal Fiyat Lojistik Global Merkez Pazarlama

17
Türkiye İlaç ve Tıbbi Cihaz Kurumu’nda Dış Paydaşların Yeri
Kurum Başkanı Özel Kalem Birimi İç Denetim Birimi Denetim Hizmetleri İlaç, Biyolojik ve Tıbbi Ürünler Koordinasyon Bilimsel ve Teknik Değerlendirme Klinik İlaç Araştırmaları Risk Yönetimi Kalite Kontrol Tıbbi Cihaz ve Kozmetik Ürünler Tıbbi Cihaz ve Etüd Proje, Yetkilendirme ve Koordinasyon Kozmetik Ürünler Koordinasyon Ekonomik Araştırmalar Mevzuat, Fiyat ve Araştırmalar Tıbbi Ürünler Tanıtım, Akılcı İlaç Kullanımı ve İlaç Tedarik Yönetimi Analiz ve Kontrol Laboratuvarları İlaç, Biyolojik ve Tıbbi Ürünler Laboratuvarı Tıbbi Cihaz Kontor Laboratuvarı Yönetim ve Destek Hizmetleri Bilişim İdari ve Mali Destek Hizmetleri 1. Hukuk Müşaviri Strateji Geliştirme Daire Başkanı Bilimsel Danışma Kurulları

18
Uyulması Gereken Belli Başlı Yönetmelikler
: 1262 Sayılı İspençiyari ve Tıbbi Müstahzarlar Kanunu : Farmasötik ve Tıbbi Müstahzar, Madde, Malzeme, Terkipler ile Bitkisel Preparatların Geri Çekilmesi ve Toplatılması Hakkında Yönetmelik : Farmasötik Müstahzarların Biyoyararlanım ve Biyoeşdeğerliliğinin Değerlendirilmesi Hakkında Yönetmelik : Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği : Beşeri Tıbbi Ürünlerin Güvenliğinin İzlenmesi ve Değerlendirilmesi Hakkında Yönetmelik : Ruhsatlandırılmış veya Ruhsatlandırma Başvurusu Yapılmış Beşeri Tıbbi Ürünlerdeki Değişikliklere Dair Yönetmelik : Beşeri Tıbbi Ürünler Ambalaj ve Etiketleme Yönetmeliği : Beşeri Tıbbi Ürünlerin Tanıtım Faaliyetleri Hakkında Yönetmelik

19
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
İmmünolojik Ürün Radyofarmasötik Radyonüklid Radyoaktif Madde Radyonüklid Jeneratör Radyonüklid Kit Radyonüklid Prekürsör Kan Ürünü TAEK Spesifik Aktivite Gümrük Birliği Alanı

20
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
Radyonüklid jeneratörler, radyonüklid kitler, radyonüklid prekürsörler, radyofarmasötikler ve endüstriyel olarak hazırlanmış radyofarmasötikler yönetmelik kapsamına alınmıştır. Veri imtiyazı ve uzman raporları kavramları yönetmelikte yer almaktadır.

21
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
Süre sınırlamaları; Başvuru dosyasının Bakanlığa ulaşmasından itibaren 30 gün içinde gerekli değerlendirme yapılarak durum başvuru sahibine bildirilir. Başvurunun eksik bulunması halinde başvuru sahibi eksikliklerini 30 gün içinde tamamlar. Eksikliklerin tamamlanarak Bakanlığa sunulmasından sonra yapılacak ikinci ön inceleme de 30 gün içinde sonuçlandırılır. İkinci ön incelemeye tabi tutulan ve eksikliği tamamlanmamış olan başvuru usulden reddedilerek sahibine iade edilir .

22
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
Bakanlık, ön incelemesi tamamlanmış eksiksiz bir ruhsat başvurusunu, başvurunun kabul edilmesini takiben 210 gün içinde sonuçlandırır. Ruhsat başvurusunun reddi halinde karar gerekçeli olarak başvuru sahibine bildirilir. Başvuru sahibinin karara 30 gün içinde yazılı olarak itiraz etme hakkı vardır. 30 gün içinde itiraz edilmediği takdirde, başvuru belgeleri sahibine iade edilir. Yapılan itiraz 90 gün içinde Bakanlık tarafından değerlendirilerek sonuç başvuru sahibine bildirilir. İtirazın değerlendirilmesi sırasında, gerekli görülür ise, başvuru sahibine sözlü açıklama ve savunma hakkı verilir. İtirazın değerlendirilmesi sonucunda çıkan karar kesindir ve bu karara itiraz edilemez.

23
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
Bakanlıkça ruhsatlandırılan aynı formül ve farmasötik şekildeki ürün için aynı gerçek veya tüzel kişiye, farklı bir ticari isimle de olsa ikinci bir yerli veya ithal ruhsatı verilemez. Ruhsat sahibinin değişmesi; Dosyanın eksiksiz olarak teslim edildiğine dair tutanak ve ruhsat sahibi değişikliği ile ruhsatı alan kişinin adı, soyadı, adresi, telefon ve faks numaralarıyla birlikte, ürünün kısa ürün bilgileri, kullanma talimatı, iç ve dış ambalajın birer örneği ve noter aracılığıyla yapılan devirlerde, söz konusu ürün için evvelce verilmiş olan ruhsatın aslı …vs gibi belgelerle başvurulur. Bakanlık, eksiksiz bilgi ve belgelerle yapılan ruhsat sahibi değişikliği başvurusunu 60 gün içinde sonuçlandırır.

24
Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği
Bu Yönetmeliğin yürürlüğe girdiği tarihten önce ithalat izni ile piyasaya arz edilen aşı, antiserum ve allerjen içeren biyolojik ürünler ve Radyofarmasötik Yönetmeliğine göre tescillendirilmiş ürünlerle ilgili gerekli değerlendirmeler yapılmak üzere ilgili kişiler, bu Yönetmeliğin yürürlüğe girdiği tarihten itibaren 1 yıl içerisinde Bakanlıkça istenilen belgeler ile ruhsat müracaatında bulunurlar. Bu süre zarfında ruhsat başvurusu yapılmayan ürünlerin ithalat izni ve tescil belgeleri geçersiz olur.

25
CTD, Common Technical Document
Ortak Teknik Doküman Kılavuzu ile Varyasyon Kılavuzları, Sağlık Bakanlığı İlaç ve Eczacılık Genel Müdürlüğü tarafından tarihi itibariyle yürürlüğe konulmuştur. CTD, üç ICH bölgesi olan Avrupa, ABD ve Japonya’daki ilaç ruhsatlandırma makamlarına yapılacak başvuruların planlı şekilde sunulmasını hedefleyen ve uluslararası alanda üzerinde uzlaşmaya varılan bir formattır.

26
CTD CTD beş modülden oluşmaktadır; Modül 1 İdari Bilgiler Modül 2 Kalite Bilgileri, Klinik Dışı ve Klinik Özetler Modül 3 Kimyasal, Farmasötik ve Biyolojik Bilgiler Modül 4 Klinik Dışı Raporlar Modül 5 Klinik Çalışma Raporları

27
Yeni Ürün Ruhsat Başvuru Dokümanları
CTD’nin parçası değil CTD 2.6 Klinik-dışı özet 2.4 Klinik-dışı genel bakış Modül 3 Kalite Module 4 Klinik-dışı çalışma raporları Module 5 Klinik çalışma raporları Modül 1 İdari Bilgiler 2.7 Klinik özet 2.3 Genel kalite özeti 2.5 Klinik bakış 2.1 İçindekiler 2.2 Giriş Modül 2 Common Technical Document (CTD) Başvuru Formatı CTD başvuru format: Modül 1: İdari bilgiler (sertifikalar vs) Modül 2: M3-5 özeti Modül 3: Kalite ile ilgili bölümler Modül 4: Klinik-dışı çalışmalar Modül 5: Klinik çalışmalar EMA Türkiye CTD formatı GMP değerlen. paralel GMP değerlen. önceden KÜB/KT Ön değerlendirme N/A Raportör tayini Ortalama onay 12 ay Ortalama onay 28 ay Regülasyon belirleyici EMA’yı takip etmekte Jenerik: M1, M2, M3 BY/BE (Modül 5) : 12 ay Orijinal: Module 1 – 5 : 24 ay
 Başvuru Formatı. CTD başvuru format: Modül 1: İdari bilgiler (sertifikalar vs) Modül 2: M3-5 özeti. Modül 3: Kalite ile ilgili bölümler. Modül 4: Klinik-dışı çalışmalar. Modül 5: Klinik çalışmalar. EMA. Türkiye. CTD formatı. GMP değerlen. paralel. GMP değerlen. önceden. KÜB/KT. Ön değerlendirme. N/A. Raportör tayini. Ortalama onay 12 ay. Ortalama onay 28 ay. Regülasyon belirleyici. EMA’yı takip etmekte. Jenerik: M1, M2, M3 BY/BE (Modül 5) : 12 ay. Orijinal: Module 1 – 5 : 24 ay.")
28
Türkiye’de Ruhsatın Tarihsel Gelişimi
2004 2005 2006 2007 2008 2009 2010 2011 2012 İthal ürünlerde paralel baş. eng. İlaca erken erişim kılavuzu Biyobenzer kılavuzu TIK yapılanması gerçekleşti Ruhsatlandırma yönetmeliği Elektronik başvuru Yapılmaya başlandı AB mevzuatına uyum amacıyla yayınlandı İlaç takip sistemi uygulanmaya başlandı Revize tanıtım yönetmeliği Ruhsat ve fiyat paralel süreçler haline geldi Endikasyon dışı ilaç kullanımı kılavuzu Sponsorluklara kısıtlama getirildi GMP sertifikası ithal yeni ürünlerde zorunlu hale geldi Farmakovijilans yönetmeliği Veri imtiyazı uygulanmaya başlandı Ruhsatlandırma yönetmeliğinde değişiklik E-CTD başvurusu zorunlu olarak uygulanmaya başladı Tip değişiklik yönetmeliği Kısa ürün bilgisi/ Kullanma talimatı uygulanmaya başlandı CTD formatı ruhsat başvurusunda Zorunlu oldu Sabit doz kombinas. veri imtiyazından muaf oldu Ambalaj yönetmeliği Biyobenzer ürün tanımlandı

29
Türkiye’de Ruhsatın Tarihsel Gelişiminin Etkileri
AB ile yönetmeliklerin uyumlaştırılması Ruhsat süreci 210 gün Yenilikçi ilaç ruhsat onaylarında farmakoekonomik veriler Jenerik – Orijinal ürün ruhsat süreçlerinde farklılaşma Ürün bilgilerinin hastaların anlayabileceği dilde (Kullanma talimatı) ambalajlara yerleşmesi Türkiye’de yüksek oranda rastlanan sahte ilaca karşı 2D barkod sistemi Bütçenin içeride kullanımına teşvik amacıyla, ithal ürünlerde GMP sertifikası şartı
 ambalajlara yerleşmesi. Türkiye’de yüksek oranda rastlanan sahte ilaca karşı 2D barkod sistemi. Bütçenin içeride kullanımına teşvik amacıyla, ithal ürünlerde GMP sertifikası şartı.")
30
Türkiye’de İthal Ürün Lansman Süreci
Ürünün önceliğine göre GMP süreci: Kategori 1 ürünleri ~ 12 ay Kategori 2 ürünleri ~ 24 ay Kategori 3 ürünleri ~ 36 ay Kategori 4 ürünleri ~ 48 ay Ürün önceliklendirme Dosya onayı GMP Denetimi GMP denetimi Denetim raporunun haz. Firma ile rapor yazışması GMP sertifikası TOPLAM SÜREÇ MİN 48 AY Ön inceleme Ana komisyon Teknik komisyon Analiz Yeni Ürün Ruhsat Süreci BY/BE komisyonu ~ 24 ay Farmakovijilans komis. KUB/KT komisyonu Ruhsatın kesilmesi Fiyat onayı Satış izni Lansman ~ 12 ay Geri ödeme onayı

31
Yenilikçi İlaç Lansman Sürecindeki Kritik Basamak GMP - Nasıl Hayatımıza Girdi?
Nisan 2009 Temmuz 2009 Aralık 2009 Mart 2010 Ruhsatlandırma Yönetmeliğinde Değişiklik: Yeni ürün başvurularında Bakanlıkça verilmiş GMP sertifikası maddesi yönetmeliğe eklendi. Ruhsatlandırma işlemleri devam eden ürünlerin onayları durduruldu. Bakanlıkça resmi açıklama yapıldı; tarihinden itibaren yeni ürün başvurularında GMP sertifikası zorunlu hale geldi. GMP sertifika talebi dosyasında sunulması gereken belgeler açıklandı

32
Yenilikçi İlaç Lansman Sürecindeki Kritik Basamak GMP - Denetim Süreci
KAPSAMI: Bitmiş ürün üretimi + Primer ambalajlama tesisi Biyoteknolojik ilaçlar (rekombinant DNA ile üretilen vs..) etkin madde üretimi Ruhsatlı ürünlerdeki üretim yeri değişiklikleri Aynı hat daha önce teftiş edilmişse, fiziksel denetime ihtiyaç duyulmuyor. EMA VE FDA’DEN FARKLI OLARAK: 210 günlük ruhsat süreci ile paralel yürütülememektedir. Başvuruda (diğer ülke otoritelerinin denetim raporu) pek çok ilave doküman zorunlu. Denetim formunda Türkiye’ye özgü ilave gereklilikler mevcuttur. Süreçlerinin paralel yürümemesi, yenilikçi ilaç sunumunda gecikmeye neden oluyor.
 etkin madde üretimi. Ruhsatlı ürünlerdeki üretim yeri değişiklikleri. Aynı hat daha önce teftiş edilmişse, fiziksel denetime ihtiyaç duyulmuyor. EMA VE FDA’DEN FARKLI OLARAK: 210 günlük ruhsat süreci ile paralel yürütülememektedir. Başvuruda (diğer ülke otoritelerinin denetim raporu) pek çok ilave doküman zorunlu. Denetim formunda Türkiye’ye özgü ilave gereklilikler mevcuttur. Süreçlerinin paralel yürümemesi, yenilikçi ilaç sunumunda gecikmeye neden oluyor.")
33
Ruhsat Sürecine GMP’nin Etkisi
’dan itibaren (GMP’nin uygulanmaya başlandığı tarih); Ruhsat ~1620 İthal ~180 Orijinal ~130 ~50 molekül Jenerik ~50 Yerli ~1440 ~40 ~20 molekül ~1400 31 AIFD Şirketinin GMP durumu* Başvurulan ürün sayısı 550* Başvurulan tesis sayısı 327 Denetlenen tesis sayısı 85 Sertifikası kesilen tesis sayısı 26 *Mart 2010-Eylül 2011 Mevcut yük göz önünde bulundurulduğunda, yeni başvuru olmaksızın tüm sertifikaların kesilmesinin en az 66 ayda tamamlanacağı (5.5 years) hesaplanmaktadır
; Ruhsat. ~1620. İthal. ~180. Orijinal. ~130. ~50 molekül. Jenerik. ~50. Yerli. ~1440. ~40. ~20 molekül. ~ AIFD Şirketinin GMP durumu* Başvurulan ürün sayısı. 550* Başvurulan tesis sayısı Denetlenen tesis sayısı. 85. Sertifikası kesilen tesis sayısı. 26. *Mart 2010-Eylül Mevcut yük göz önünde bulundurulduğunda, yeni başvuru olmaksızın tüm sertifikaların kesilmesinin en az 66 ayda tamamlanacağı (5.5 years) hesaplanmaktadır.")
34
Etiket Herhangi bir ürünün birincil kabı üzerinde bulunan tüm yazılı, basılı veya grafik materyal veya bir tüketici ürününe ya da herhangi bir tüketici ürünü içeren bir ambalaja iliştirilmiş veya bu ambalaj üzerinde görülen bu türden materyal sunumudur.

35
Beşeri Tıbbi Ürünler Ambalaj ve Etiket Yönetmeliği*
Dış Ambalaj 30/07/2004 tarihli ve sayılı resmi gazetede yayımlanan Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliği gereğince ambalajların üzerinde geri kazanılabilir ambalaj sembolü ve ambalajın cinsine ait numara ve kısaltma bulundurulur. Son kullanma tarihi (gün) ay ve yıl olacak şekilde yazılır. *AB ile tam uyumlu olarak hazırlanmıştır.
 ay ve yıl olacak şekilde yazılır. *AB ile tam uyumlu olarak hazırlanmıştır.")
36
Beşeri Tıbbi Ürünler Ambalaj ve Etiket Yönetmeliği*
İç Ambalaj Ruhsat/izin sahibinin ismi veya amblemi Son kullanma tarihi (gün) ay ve yıl olacak şekilde yazılır. *AB ile tam uyumlu olarak hazırlanmıştır.
 ay ve yıl olacak şekilde yazılır. *AB ile tam uyumlu olarak hazırlanmıştır.")
37
Beşeri Tıbbi Ürünler Ambalaj ve Etiket Yönetmeliği
Prospektüs Kullanma Talimatı Ürüne ait KÜB’e uygun olarak KULLANICININ kolay anlayabileceği şekilde hazırlanır* Ürünün tanımlanması kısmında ürünün bebeklere, çocuklara veya erişkinlere yönelik olduğuna dair bilgi Kullanma talimatının en son güncellendiği tarih * Beşeri Tıbbi Ürünlerin Ambalaj Ve Hasta Kullanma Talimatının Okunabilirliğine İlişkin Kılavuz

38
Kısa Ürün Bilgisi (KÜB)
Etiket ve KÜB farklı kavramlardır. KÜB her zaman bir ürüne eşlik eden yazılı, basılı veya görsel materyaldir. KÜB, reçeteleme bilgileri veya prospektüs anlamına da gelir. KÜB’nin amacı, sağlık hizmeti uzmanlarına ilaçları doğru şekilde reçete etmeleri için ihtiyaç duydukları bilgileri sağlamaktır. KÜB, hekimler ve eczacılar tarafından kullanılan önemli bilgilerin temel kaynağıdır.

39
KÜB’ün 7 Ana bölümü 1- Endikasyonlar ve Kullanım
Bu bölüm, ilacın ne için endike olduğunu ve bir ilaç kullanmanın sınırlamalarını belirtir. 2- Dozaj ve Uygulama Bu bölüm önerilen ilaç rejimlerini ve tüm özel uygulama talimatlarını belirtir. 3- Kontrendikasyonlar Bu bölüm ilacın kesinlikle KULLANILMAMASI gereken durumları belirtir. 4- Uyarılar ve Önlemler Bu bölüm, advers reaksiyonları önlemek veya hafifletmek üzere yapılabilecek izleme veya test etme gibi faaliyetler hakkında bilgiler içerir. İlaçtan kaçınması gereken hastalar da buraya eklenir.

40
KÜB’ün 7 Ana bölümü 5- Advers Reaksiyonlar
Bu bölüm, en yaygın şekilde oluşan advers reaksiyonları ve bu oluşumların yüzdelerini belirtir. 6- İlaç Etkileşimleri Bu bölüm, klinik olarak önemli ilaç etkileşimlerini ve bunları önlemeye yönelik talimatları belirtir. 7- Özel Popülasyonlarda Kullanım Bu bölüm özel durumlarda ve koşullarda ilaç kullanımını açıklar. Gebelik, doğum, emziren anneler, pediyatrik, geriyatrik ya da karaciğer veya böbrek yetmezliği olan hastalar gibi.

41
KÜB’ün Kalan 7 bölümü 1- Doz aşımı
Bu bölümde, doz aşımı belirtileri ve semptomları açıklanır. 2- Klinik Farmakoloji Bu bölümde ilacın kimyasal adı ve formülü açıklanır. 3- Klinik Dışı Toksikoloji Bu bölümde ilacın karsinojenik veya mutajenik potansiyeli açıklanır. 4- Klinik Çalışmalar Bu bölümde, onaylanmış endikasyonlar için ilacın güvenli ve etkili kullanımı destekleyen klinik çalışmalar açıklanır.

42
KÜB’ün Kalan 7 bölümü 5- Dozaj Formları ve Kuvvetleri
Bu bölümde, ilacın mevcut dozaj formları ve kuvvetleri belirtilir. 6- Açıklama Bu bölümde, insanlarda klinik farmakoloji açıklanır. 7- Tedarik Şekli/Saklama ve Kullanım Bu bölümde üretilen birimler ve ambalajlar ve ilacın kullanma ve saklama talimatları açıklanır.

43
2006 yılında ABD’de KÜB’e 2 yeni bölüm eklenmiştir
İçindekiler Tablosu Önemli Noktalar belgedeki diğer bölümlere çapraz referans verir ve ayrıca son ana değişiklikler, tüm uyarı kutuları ve ilk onay tarihini içerir. Uyarı Kutusu, ABD KÜB’sine özgüdür. özellikle ölüme ya da ciddi yaralanmaya neden olabilecek olan ciddi uyarılar için gereklidir.

44
CCDS, Company Core Data Sheet
Şirket Çekirdek Veri Sayfası, şirketin ilaca dair tıbbi ve bilimsel bilgilerini temsil eder. İlacın pazarlanması planlanan her bir ülke için yerel KÜB’ler geliştirmekte kullanılır.

45
Danışmanlarla Etkileşim
Yeni ilaç geliştirme sırasında danışmanlarla sürekli bir diyalog sağlamak önemlidir. Klinik bir çalışmada ne işe yarar ve ne yaramaz? Karşılanmamış tıbbi ihtiyaçlar nelerdir? Mevcut tedaviyle ilgili temel endişeler nelerdir?

46
NDA, New Drug Application
NDA incelendikten sonra, FDA, Tam Cevap Mektubu Tam cevap mektubu tarafından verilen endikasyon, bir başvuru için inceleme döngüsünün tamamlandığı ve başvurunun onaya hazır olmadığıdır. Onay Mektubu, onaylanmış etiketlemeyi içerir.

47
BEDEL ? Çoğu ülkede bir pazarlama başvurusunun gönderilmesi ve incelenmesi ücrete tabidir. ABD’de bu ücrete ―Kullanıcı Ücreti adı verilir yılında sunulan bir başvuru için, tam klinik verileri içeren yeni ilaç başvurusunun kullanıcı ücreti 1,405,500 dolardır.

48
EKSİK BAŞVURU Şirket FDA'dan bir ―Dosya Reddi kararı almışsa, dosyalama sırasında sunulan başvuru ücretinin yarısı kadarlık bir ceza hesaplanır ücret planına dayanarak, başvuruda %50 peşinatın yarısı ceza olarak verilir, Bu da şirketin bir milyon doların üçte birinden daha fazla ceza alacağı anlamına gelir!

49
TEŞEKKÜR Bazı Slaytlar,
İpek Altmışoğlu / MSD Türkiye Ruhsatlandırma Müdürü /13 Haziran 2012/ İstanbul / İlaç Keşfi ve Geliştirilmesi Programı Eğitim Toplantısı’ndan alıntıdır.

50
DAHA FAZLA BİLGİ İÇİN

Benzer bir sunumlar




 Bu Yönetmeliğin amacı, kamu kurum ve kuruluşları hariç olmak üzere ondan az çalışanı bulunanlardan, tehlikeli ve çok.>")














